










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCTION
SARS-CoV-2 is remarked as a novel virus responsible for the pandemic of the severe acute respiratory syndrome, also known as Coronavirus Disease 2019 (COVID-19). First discovered in December 2019, SARS-CoV-2 has been the causative agent for a pneumonic illness initially detected in Wuhan City, Hubei province, China [1, 2, 3, 4, 5, 6, 7]. As of 31 May 2020, more than 6.1 million cases have been reported across 187 countries and territories, resulting in more than 371,000 deaths [8]. More than 1.7 million people have recovered. Common symptoms include fever, cough, fatigue, shortness of breath, and loss of smell and taste. At the same time, most cases result in mild symptoms, some progress to viral pneumonia, multi-organ failure, or cytokine storm [8].
Cytokine Storm: This is described as severe acute inflammation that arises from an exaggerated immune response to the presence of the SARS-CoV-2, which is considered as one of the most critical negative prognostic markers in COVID-19 disease [9, 10, 11, 12]. “If there is a cytokine storm, the use of selective cytokine blockade agents (e.g., anakinra or tocilizumab) is thought to be helpful. Anakinra is one of the interleukin antagonists, and it blocks the effect of interleukin-1, which is highly increased during the cytokine storm” [9, 11]. “Strategies from China's National Health Commission comprise the IL-6 inhibitor tocilizumab for severe COVID-19 cases and high IL-6 levels during the cytokine storm phase of COVID-19 disease, which is being assessed in clinical trials” [9, 10, 11, 12].
Dexamethasone Discovery: On 16 June 2020, researchers at the University of Oxford announced that they had identified the first drug proven to reduce mortality from COVID-19 [14]. “Dexamethasone, a cheap steroid, reduces deaths by a third among the most severely ill patients who have needed mechanical ventilation and a fifth among critically ill patients who have required oxygen support. It is set to become the standard of care across Britain,” [14]. “Clinicians globally will, undoubtedly, follow suit. Dexamethasone is an anti-inflammatory that is already used to treat a variety of health problems, such as rheumatoid arthritis, eczema, asthma, and some cancers” [14]. “It is important to note that even though dexamethasone has no documented antiviral capabilities; but, it was included in the randomized trial because steroids and mainly naturally occurring corticosteroids were included in the SARS testing with mixed results” [14]. Besides, there are concerns from scientists about accentuation of the side-effects associated with steroid use, especially with the use of Dexamethasone for COVID-19 patients [15, 16].
COVID-19 Pandemic Updates: Other author reports that “the COVID-19 Pandemic is swirling the world with over 8,000,000 confirmed cases, 440,000 deaths, and about 10.2% needing support in form of Mechanical Ventilators and oxygen” [8]. However, “most people who fall sick with COVID-19 will experience mild to moderate symptoms and recover without special treatment. However, the number of death from COVID-19 related conditions is on the increase hence the need to find a lasting solution,” [8].
Aspirin Story: Furthermore, ever since Felix Hoffman, in 1897, discovered a new technique for acetylating the phenolic hydroxyl group of salicylic acid to form acetylsalicylic acid (Aspirin), the world has not remained the same [17]. Interestingly, “Hoffman developed a method to retain the analgesic and antipyretic properties of salicylic acid while decreasing the side effects associated with prolonged administration. Also, the utilization of aspirin derivatives dated back as over 2000 years ago. During these periods, salicylates were initially derived from plant extractions,” [17]. Besides, “salicylic acid, in particular, was isolated from the Willow tree. By the early 1900s, the therapeutic benefits of Aspirin (and its salicylate) were widely recognized. Over time, other drugs were developed with the same anti-inflammatory, analgesic, and antipyretic activities,” [17]. “Aspirin has been used for over a century to treat the cardinal signs of inflammation (heat, redness, swelling, and pain). In contrast, recently, it has been shown to prevent intravascular thrombosis and cancer and slow Alzheimer's disease,” [17].
OBJECTIVES OF THIS STUDY
We highlight principal mechanisms by which Aspirin inhibits acute inflammation and alters platelet-biology; therefore, hypothesized that Aspirin might prove highly beneficial as a novel therapeutic drug for combating severe acute inflammation and thrombosis associated with the cytokine storm in COVID -19 patients. The communiqué also suggests possible strategies for maximizing the gain of Aspirin as a wonder-drug of the future.
HISTORICAL BACKGROUND
About 400BC, Hippocrates recommended a brew of leaves from the willow tree (Cortex salicis) to ease pain in childbirth. This story is the first recorded mention of the use of salicylates for pain relief. Salicylates are widely distributed in plants, and many herbal remedies probably depend on them for their effect [18, 19]. In 1763, the Reverend Edward Stone wrote to the President of the Royal Society in London, reporting that he had successfully used a powder prepared from the bark of the typical white willow to treat fever in “over 50 patients suffering from various agues” [18, 20]. The Reverend Stone's letter brought salicylates to chemists' attention, and, in 1859, salicylic acid was first synthesized from carbolic acid. It was used to relieve pain and fever, although it proved to be irritating to the stomach. Because of its irritant action, chemists made efforts to find a derivative of salicylic acid that could be better tolerated [18, 20]. In 1897, Felix Hoffman, working in a laboratory owned by Friedrich Bayer in Germany, synthesized acetylsalicylic acid. Hoffman and Bayer gave the name aspirin to the new preparation — A for acetylation and spirin, part of the name for meadow-sweet (Spiraea ulmaria), a plant rich in salicylates. Bayer patented the name and began marketing the powder in 1899. It was a huge success, and sales increased. The production and marketing of Aspirin laid the foundations of the modern pharmaceutical industry, and Friedrich Bayer and Company is generally acknowledged to be the first pharmaceutical company. Aspirin was accepted with great enthusiasm, and has justifiably been called the first miracle drug [18, 21].
INDICATIONS FOR ASPIRIN
The traditional primary use was for relief of pain and reduction of fever. Still, it was also recommended for use in rheumatoid arthritis, migraine, inoperable cancer, gout, rheumatic fever, acute tonsillitis, corns, and warts [18, 21]. Aspirin is still the gold standard against which other analgesics, anti-inflammatory, and fever-reducing drugs have been measured. Indeed, Aspirin's success stimulated the search for newer, more potent, and safer products — nonsteroidal anti-inflammatory drugs (NSAIDs). Some of these are more potent than Aspirin, some are safer, and some (the recently introduced cyclooxygenase-2 [COX-2] inhibitors), are more specifically targeted at an enzyme level. The common cyclooxygenase pathway is as highlighted in Figure 1.
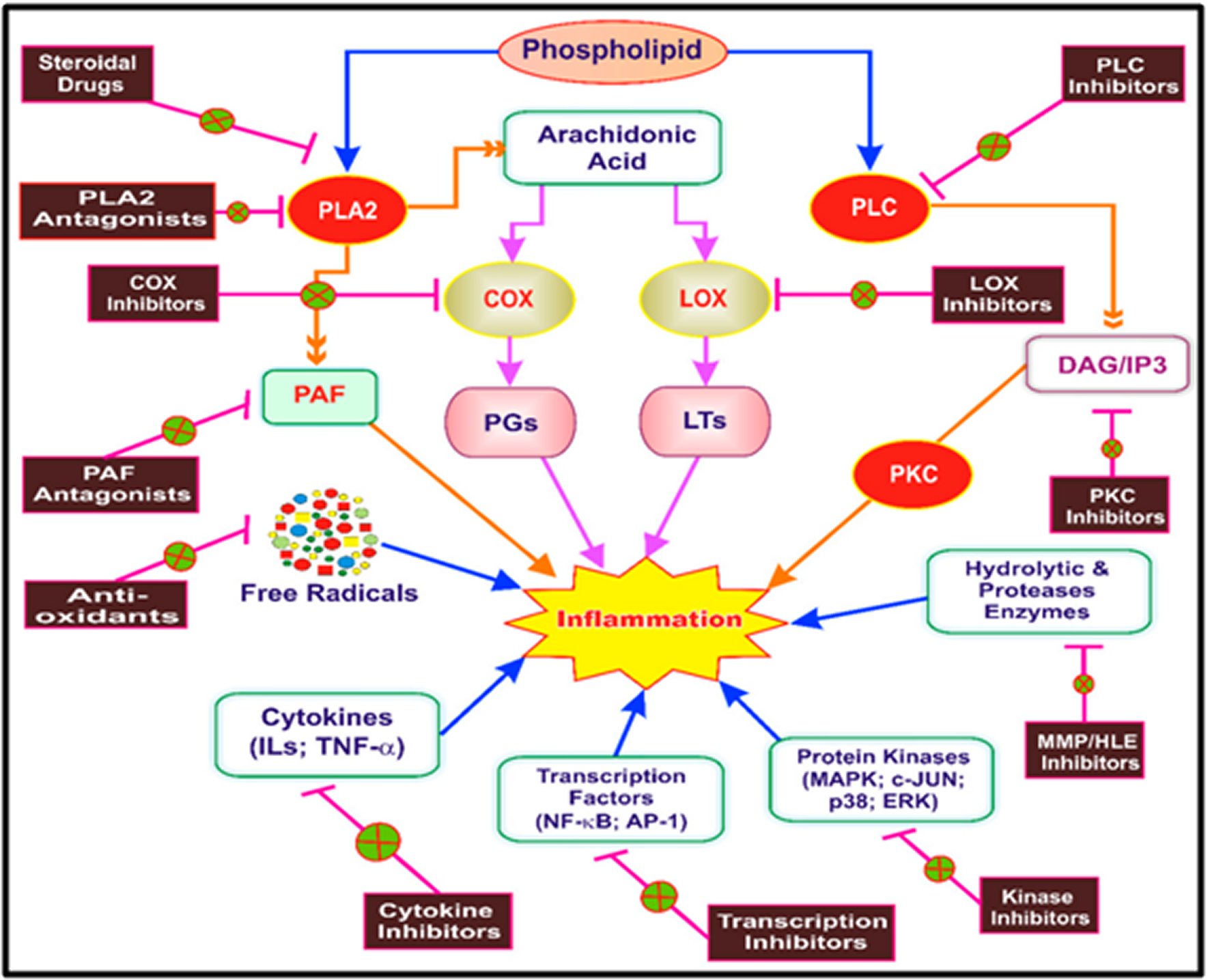
Figure 1. The common cyclooxygenase pathway.Key targets for initiation of inflammation. The arrows in the figure represent the process and fork represents inhibition of target. PLA2: Phospholipase A2; PLC: Phospholipase C; PKC: Protein kinase C; PAF: Platelet-activating factor; DAG: Diacylglycerol; IP3: Inositol triphosphate; COX: Cyclooxygenase; LOX: Lipoxygenase; PGs: Prostaglandins; LTs: Leukotrienes; MMP: Matrix metalloproteinase; HLE: Human leukocyte elastase; ILs: Interleukins; TNF-α, Tumor necrosis factor-alpha; NF-κβ: Nuclear factor kappa beta; AP-1: Activator protein-1; MAPK: Mitogen-activated protein kinase; P38: P38 kinase; c-JUN: c-Jun N-terminal kinase; ERK: Extracellular signal-regulated kinase.
Nevertheless, Aspirin remains a mainstay of analgesia. Of great interest is a marked reduction in colorectal cancer in habitual aspirin takers. This finding has been reported in many studies [18, 22, 23]. Low-dose Aspirin reduces the risk of a cardiovascular event, such as heart attack, stroke, or deep vein thrombosis, by about 30 percent [18]. Aspirin equally reduces the risk of a cardiovascular event associated with Kawasaki disease [24, 25, 26]. It has been more widely distributed than any other drug — Scott took it to the Antarctic, Hillary to Everest, and the astronauts to the moon [18, 19, 20, 21].
A NOVEL MECHANISM OF ACTION FOR ASPIRIN
CYCLOOXYGENASE (COX) PATHWAYS
Aspirin is a widely used nonsteroidal anti-inflammatory drug (NSAID). The common cyclooxygenase pathway is as highlighted in Figure 1 [18, 19, 20, 21]. It is well documented that “Aspirin irreversibly inhibits cyclooxygenase (COX) by acetylation of an amino acid serine residue, as seen in Figure 2. Also, it blocks the subsequent biosynthesis of prostaglandins and thromboxane,” [27]. “COX has at least two forms, COX-1 and COX-2. COX-1 is the main form present in mature platelets in the blood, where it transforms arachidonic acid to the intermediates PG-G/H, which are subsequently converted to thromboxane A2. Thromboxane A2 is a vasoconstrictor and potent platelet activator,” [27]. Thus, inhibition of thromboxane A2 formation explains Aspirin's antithrombotic properties, as seen in Figure 3. In the early 1990s, the second form of COX was identified, namely, COX-2. COX-2 was initially conceptualized as an “inducible” COX that is elevated in its quantity by a wide range of agents that stimulate inflammation or cell division and seems to be responsible for local formation during inflammation and cancer [27, 28].
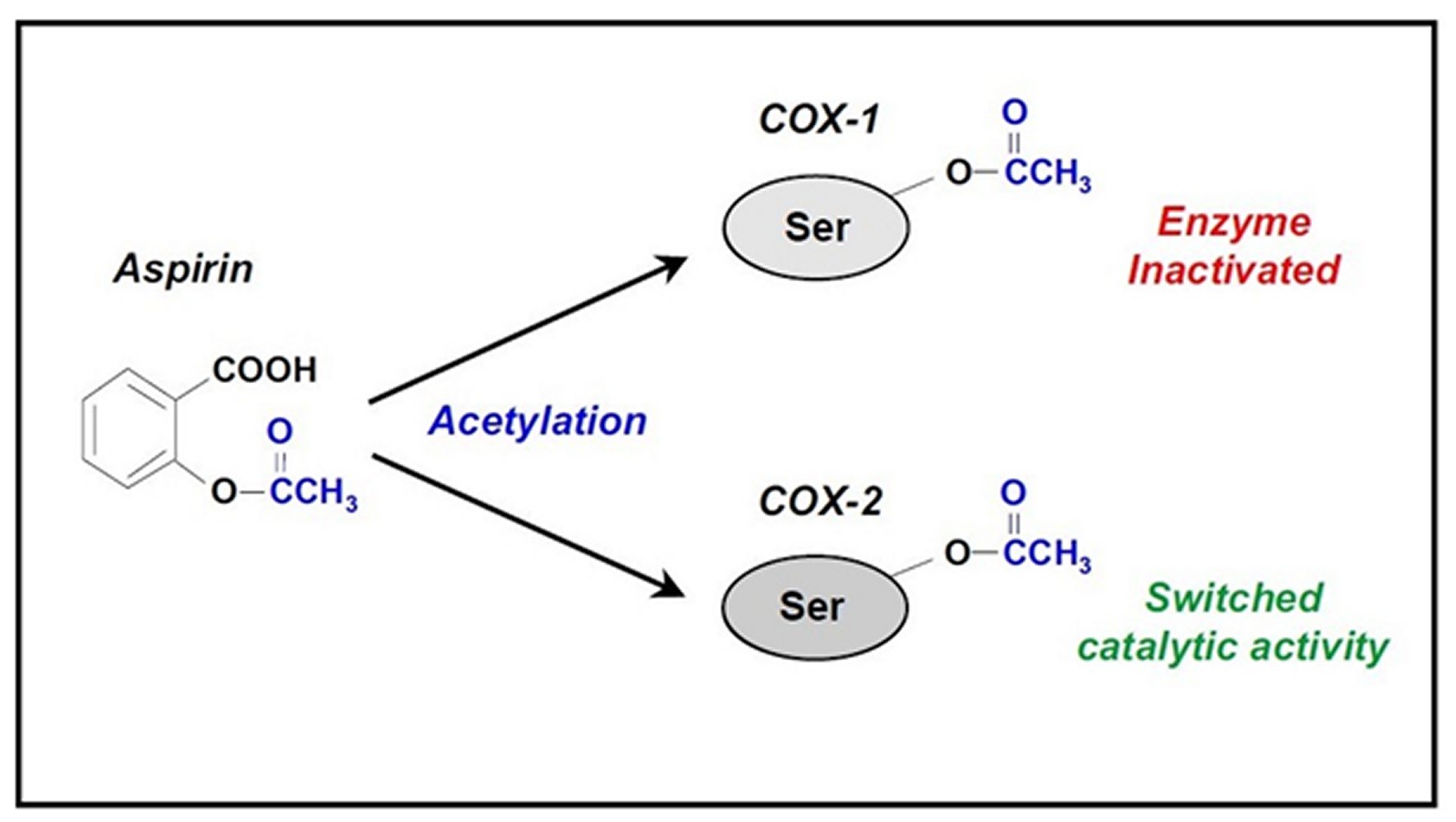
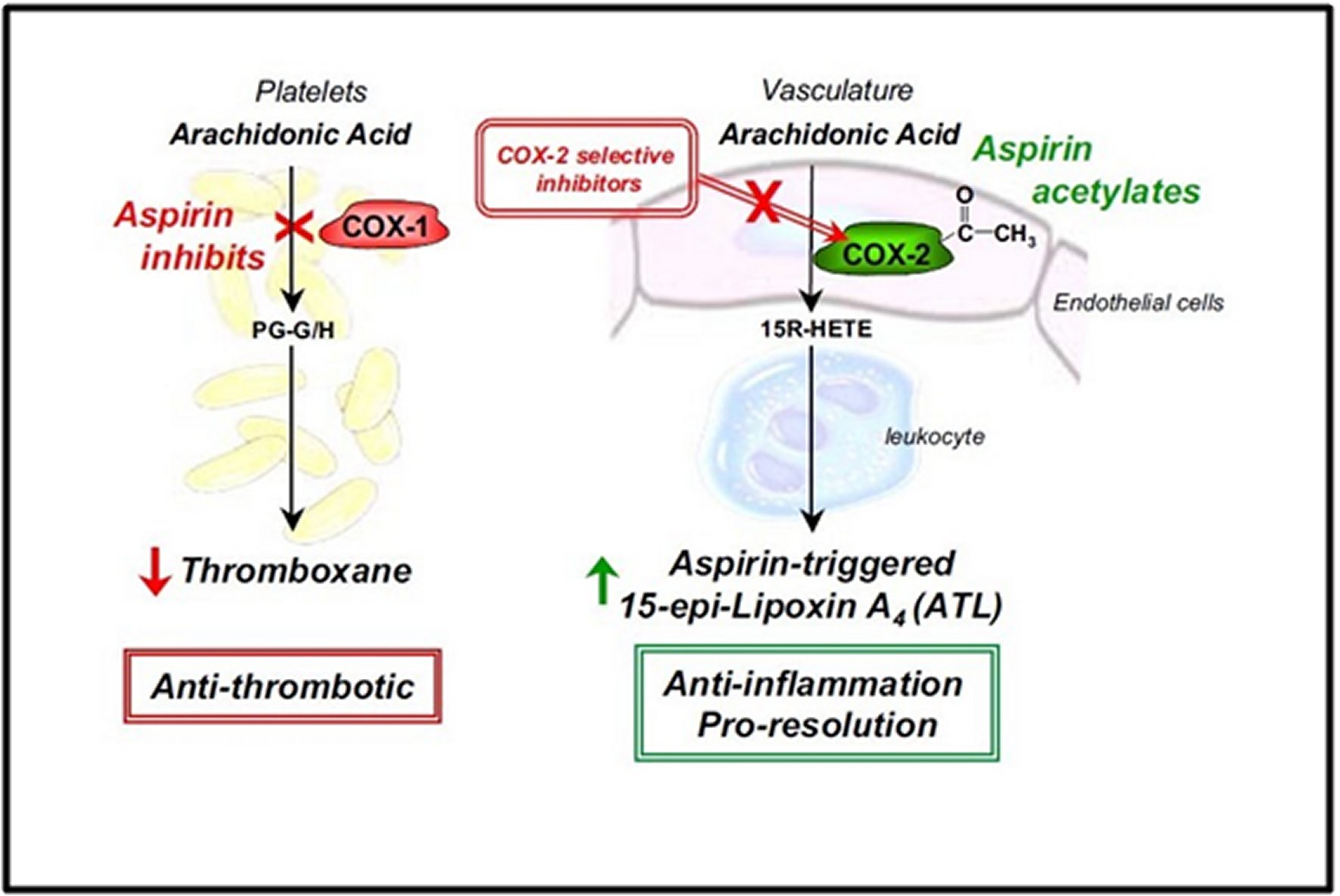
Many recent studies demonstrated that “low-dose aspirin evokes beneficial effects not only in the prevention and treatment of cardiovascular diseases, but also in decreasing the incidence of lung, colon and breast cancers, and perhaps Alzheimer's disease” [27, 29]. “Although inhibition of prostaglandins and thromboxanes can account for aspirin's therapeutic benefits, aspirin's ability to regulate neutrophil-mediated inflammation remains of interest,” [27, 29]. Hence, low-dose aspirin effects that go beyond the inhibition of prostaglandins and thromboxanes are becoming increasingly apparent. In this regard, the report found statistically significant ability of Aspirin to inhibit prostaglandins and thromboxanes, can also 'switch on' the production of the body's anti-inflammatory lipid mediators, namely aspirin-triggered lipoxins (ATL) [27, 29]. “This novel class of mediators functions as local ‘braking signals' in inflammation and actively participates in dampening host immune responses and quickly bringing the inflammatory reaction to a closure, a process called resolution. Thus, they may account at least in part for Aspirin's clinical benefits distinct from Aspirin's antithrombotic action,” [27, 29].
ATL BIOSYNTHESIS VIA ASPIRIN-ACETYLATED COX-2
“This novel aspirin action involves cell-cell communication between COX-2-bearing cells such as vascular endothelial cells or epithelial cells and their interaction with leukocytes in a process known as transcellular biosynthesis seen in Figure 4” [27, 30]. Briefly, “aspirin acetylates add an acetyl group to COX-2 and redirects COX-2's catalytic activity away from generating intermediates of prostaglandins and thromboxanes and towards producing another compound (15R-HETE)” [27, 30]. “This latter compound is converted to a different compound (15-epi-Lipoxin A4) by 5-lipoxygenase in activated neutrophils and then rapidly released. The new compound is termed aspirin-triggered 15-epi-lipoxin A4 (ATL)” [27, 30]. It is noteworthy that the ability to initiate ATL formation is unique to Aspirin, as other widely used NSAIDs of general COX inhibitors are unable to generate ATL [22, 25]. Along these lines, recent findings from laboratory findings demonstrated that “in addition to arachidonic acid, the omega-3 polyunsaturated fatty acids (found in fish oil), namely eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), can also interact with aspirin-acetylated COX-2 to generate bioactive compounds, namely resolvins” [27, 29]. Therefore, Aspirin converts COX-2 into a 'protective mediator-generating system' that produces arrays of novel mediators with anti-inflammatory and pro-resolving properties.
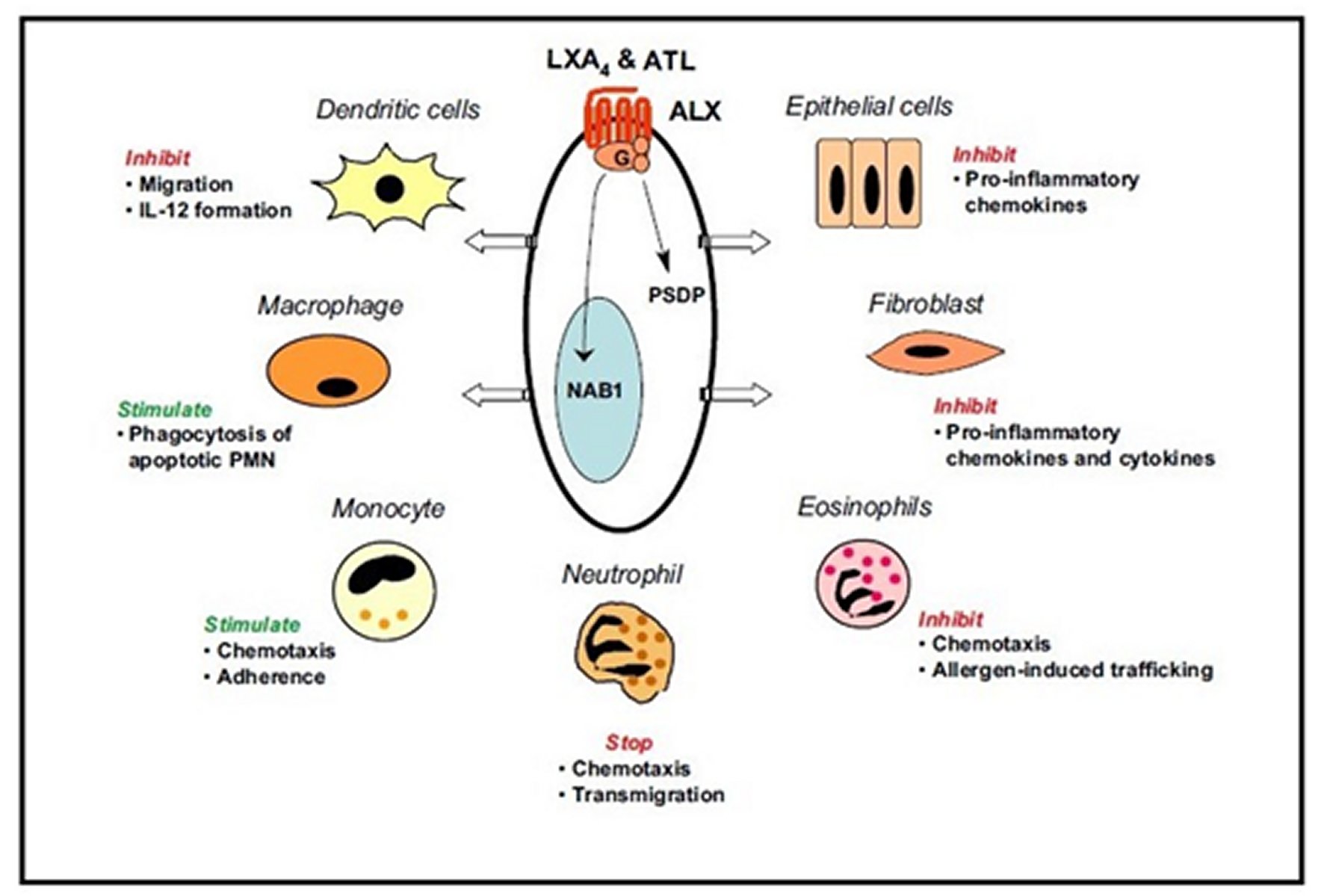
BIOACTION AND FORMATION OF LIPOXIN A4 AND ATL
“Lipoxin A4 and ATL were the first to be recognized as endogenous anti-inflammatory lipid mediators, displaying similar biological actions in regulating leukocyte activation. In this manner, these compounds regulate essential initial steps in tissue inflammation and its resolution,” [27, 29]. “Both Lipoxin A4 and ATL act at the low nanomolar concentration range via the interaction with a specific cell surface receptor (denoted the Lipoxin A4 receptor) in various cellular systems, as seen in Figure 4,” [27, 29]. Besides, they possess potent protective actions in a variety of experimental animal models of disease, including peritonitis, dermal inflammation, reperfusion injury, periodontitis, asthma, and angiogenesis, etc., [27, 29]. “Using a newly developed specific bioassay (enzyme-linked immunosorbent assay, ELISA) and mass spectrometry systems, ATL generation has been documented during cell-cell interactions and in a variety of animal models” [27, 31].
ATL FORMATION IN A RANDOMIZED HUMAN TRIAL
“ATL has anti-inflammatory actions in experimental animal models, and the protective effect of ATL is likely to provide alternate explanations for Aspirin's therapeutic impacts. Therefore, the researchers sought to determine whether Aspirin, administered in standard clinical doses to healthy volunteers, would initiate anti-inflammatory ATL generation and its relationship to Aspirin's antiplatelet activity (i.e., thromboxane formation) in a randomized human trial” [27, 32]. “A randomized, double-blind, and placebo-controlled clinical trial was conducted involving three different doses of Aspirin (81 mg, 325 mg, and 650 mg), taken once daily in the morning over eight weeks. These are the most frequently administered aspirin doses in the United States and are clinically recommended for different therapeutic purposes. They are as follows, i) Low dose (81 mg) as antithrombotic for long-term prevention of cardiovascular events, ii) Medium dose (325 mg) for acute situations such as myocardial infarction (heart attack) and thrombotic stroke, and iii) A higher dose (650 mg) for relieving pain and antipyretic (anti-fever) effects. Blood samples were collected before and after eight weeks of Aspirin or placebo dosing, and thromboxane B2 (TXB2, a stable metabolite of TXA2) and ATL levels were determined” [27, 32]. “The researchers found that in the low-dose 81 mg aspirin group, ATL levels at eight weeks are significantly higher than those before aspirin treatment. In parallel determinations, TXB2 levels decreased significantly after eight weeks of treatment. In sharp contrast, in the placebo group, neither ATL nor TXB2 levels at 8-weeks are substantially different from before administration,” [27, 32]. “This report is the first documentation in a randomized human trial of healthy volunteers that Aspirin increases anti-inflammatory ATL as well as inhibits prothrombotic thromboxane. Along these lines, we also assessed Aspirin's 'net' impact by determining the 'net change' in ATL and TXB2 levels (i.e., ATL minus TXB2) for each randomized dose group” [27, 32]. Statistical analysis indicated that this “net change" in each aspirin dose group is significant compared to the placebo group. While ATL production in humans is dependent on low-dose daily Aspirin (81 mg), the magnitude of this effect at higher doses (325mg or 650 mg of daily Aspirin) is not further enhanced above that observed with the 81 mg aspirin daily dose [27, 32]. “These observations are of clinical interest, as numerous recent reports indicated that the beneficial effects of high-dose aspirin (500 to 1500 mg daily) on reducing the incidence of vascular events do not exceed that already achieved with lower doses (75 to 150 mg daily)” [27, 32]. Thus, “given the vital contribution of inflammation in cardiovascular diseases and the anti-inflammatory and pro-resolving properties of ATL; within the vasculature, local ATL formation is likely relevant in the cardioprotective actions of Aspirin documented in many clinical studies. ATL may protect against the initial inflammatory events that could lead to harmful cardiovascular effects. Therefore, ATL could also serve as a simple clinical parameter to verify an individual's anti-inflammatory response with low-dose aspirin treatment,” [27, 32].
ATL AND COX-2 INHIBITORS
“COX-2 is present in many cell types, including megakaryocytes (precursor cells to mature platelets) and vascular endothelial cells. COX-2 is expressed following exposure to laminar shear (damage to the extracellular matrix, which cells use as a scaffold). It is likely that COX-2 is constitutively shown in blood vessel walls and promotes local ATL production in healthy subjects taking low-dose Aspirin,” [27, 32].
“COX-2 selective inhibitors block prostacyclin (prostaglandin I2) production in the vasculature without affecting platelet COX-1 and thromboxane production. Prostacyclin is a vasodilator and interferes with platelet reactivity. Hence, an individual treated with COX-2 inhibitors could be more 'prothrombotic' since platelet thromboxane production continues when COX-2 selective inhibitors are given” [27, 32, 33]. In this regard, it is essential to note that COX-2 inhibitors prevent ATL formation during leukocyte-endothelial cell interactions and by the inflamed gastrointestinal tract of rats [27, 32, 34]. Thus, it is also likely that COX-2 inhibitors may “temper” the beneficial impact of low-dose Aspirin by blocking the body's natural anti-inflammatory ATL formation in humans.
15-EPI-LIPOXIN A4-MEDIATED INDUCTION OF NITRIC OXIDE
Paul-Clark MJ et al., in their study, found that the inhibition of prostaglandin synthesis, as described above, has never fully explained Aspirin's repertoire of anti-inflammatory properties [35]. In their further study found in acute pleuritis that Aspirin, but not salicylate, indomethacin, or piroxicam, increased plasma nitric oxide (NO), which correlated with a reduction in inflammation. Inhibiting aspirin-elicited NO pharmacologically in this model nullified the anti-inflammatory effects of Aspirin. Moreover, Aspirin was not anti-inflammatory in either constitutive (eNOS) or inducible NO synthase (iNOS) knockout mice with IL-1–induced peritonitis. It transpires that Aspirin generates NO through its unique ability to trigger the synthesis of 15-epi-lipoxin A4 [35]. Aspirin and 15-epi-lipoxin A4 were shown to inhibit leukocyte trafficking in a NO-dependent manner using intravital microscopy on IL-1–stimulated mouse mesentery. Not only did Aspirin inhibit leukocyte-endothelial interaction in a manner similar to NO in wild-type mice, but both Aspirin and 15-epi-lipoxin A4 had markedly reduced effects on leukocyte-endothelial cell adherence in eNOS- and iNOS-deficient mice compared with wild type [35]. Collectively, these data suggest that Aspirin triggers the synthesis of 15-epi-lipoxin A4, which increases NO synthesis through eNOS and iNOS. This aspirin-elicited NO exerts anti-inflammatory effects in the microcirculation by inhibiting leukocyte–endothelium interactions [35].
PATHOPHYSIOLOGY OF SARS-COV-2 INFECTION
PATHOPHYSIOLOGY OF SARS-COV-2 INFECTION
PHASE ONE: MILD CATEGORY
“Once the virus makes its way to the respiratory tract, it reaches the alveoli, where their primary targets are the type-2 pneumocytes [36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46]. Type-1 pneumocytes play a role in gas-exchange,” [36, 47]. In contrast, as the type-2 pneumocytes produce an essential substance called surfactant (a substance that reduces the surface tension within the alveoli and contributes to the lungs' elastic properties, thereby making breathing relatively easy and effortless) [36, 48, 49]. “Type-2 pneumocytes are the primary targets of the virus which infects the cells and initiates its multiplication inside the body. The S-spike of the virus binds with the angiotensin-converting enzyme 2 (ACE-2) on the surface of the pneumocytes type-2 cells” [36, 50]. “ACE-2 is a member of the ACE family and a type-I transmembrane metallocarboxypeptidase and a regulator of the Renin-Angiotensin system and hence an essential target for management of hypertension. It is distributed in many tissues in the body, including lung, kidney, gastrointestinal (GI) tract, and cardiovascular system” [36, 51, 52, 53, 54, 55, 56, 57, 58]. This process might explain the systemic manifestations, renal compromise, GI dysregulation, cardiovascular dysfunction, and multi-organ failure in certain SARS-CoV-2 cases.
Interestingly, many of the COVID-19 cases present with extrapulmonary symptoms. This presentation is because ACE-2 is expressed in many cell types in the body. ACE-2 mediated functions imbibe a wide array of physiological events that can serve as an integrative model to reconstruct the COVID pathology. In elderly individuals (those more than 60 years of age) and those with diabetes or coronary artery diseases, COVID-19 has been reported to lead to serious cardiac complications (like heart failure) in some instances. Acute liver and kidney failures also reported as complications in some (but not all) infected individuals [36, 51, 52, 53, 54, 55, 56, 57, 58]. “Gastrointestinal related symptoms (like diarrhea, nausea, and vomiting) are commonly present, and sometimes the only presenting features in some infected individuals. This finding is where ACE-2 mediated pathology of COVID-19 finds additional justification. The binding of SARS-CoV-2 to ACE-2 may potentially explain these extrapulmonary symptoms in COVID-19” [36, 51, 52, 53, 54, 55, 56, 57, 58].
Furthermore, RNA viruses may act through activation of endogenous reverse transcriptase to archive their genome in the host genome in the form of DNA [36, 59, 60], but that does not seem to be the case with SARS-CoV-2. The preceding action suggests the reason that antiretroviral therapy is thought to have the potential to treat COVID-19 patients. Once the RNA is released into the cytoplasm of type-2 pneumocytes, it then acts as a messenger RNA [36, 59, 60]. It utilizes the host cell ribosome machinery to translate the polyproteins, which are cleaved by specific enzymes (proteases) into active protein particles. Including the S-spike that are substrates against which antibodies are being designed for therapeutic/vaccination purposes [36, 61, 62, 63, 64], the capsid, and various other protein moieties [36, 65, 66] requisite for the assemblage of the active and viable virus [36, 67, 68, 69, 70, 71, 72]. “It appears that certain protease-inhibitors can have the potential to inhibit this step and, therefore, may act as potential drugs for COVID-19 treatment [36, 73]. Ritonavir, a potent protease inhibitor (which has been used to treat HIV infection), may serve as a potential candidate for such investigations” [36, 74, 75, 76]. “One of the translated proteins of SARS-CoV-2 is the RNA-dependent-RNA polymerase, which leads to the synthesis of multiple RNA copies (which are the genome of the virus). Given enough copies of the RNA being produced within the cytoplasm of the host cells (which in this case are pneumocytes), and enough proteins essential for the viral structures, these two moieties combine to give rise to hundreds and thousands of viral particles within the cell” [36, 77]. Once that occurs, the cell either activates its apoptotic mechanism or is being destroyed by the macrophages. This process leads to the release of the multiple copies of the viable virus into the extracellular matrix and interstitial fluid. The new amplified progeny of the virus now has the opportunity to infect other types-2 pneumocytes. Given enough viruses being produced, they affect type-2 pneumocytes of other alveoli and enter the blood circulation to invade different cell types (particularly those expressing ACE-2). Then the coronavirus mediated pathophysiology [36, 78] potentially leading to pneumonia begins. Once type-2 pneumocyte gets ruptured, it subsequently attracts macrophages through the production of inflammatory mediators. The so activated macrophages [36, 79, 80, 81, 82] could then secrete specific cytokines, including interleukin 1 (IL1), interleukin 6 (IL6), and tumor necrosis factor-alpha (TNF-α). These so released cytokines mediate their effects through moving into the bloodstream and immediately causing systemic inflammatory response syndrome (SIRS) [36, 79, 80, 81, 82]. “The American College of Chest Physicians and Critical Care Medicine in 1991 defined SIRS as a clinical syndrome characterized by systemic inflammation and widespread tissue injury. SIRS is defined by several clinical variables including temperature >38°C or <35°C, heart rate >90 beats/min, respiratory rate >20 breaths/min or PCO2 < 32 mmHg, and WBC > 12000 cells/mm3 or <4000 cells/mm3” [36, 79, 80, 81, 82].
“The dilation of blood vessels characterizes SIRS by acting on endothelial cells of the microvasculature around the alveoli. Dilation of the smooth muscles of the alveolar microvasculature can lead to an increase in the permeability of the blood vessels around the alveoli. One of the parallel events in this is the endothelial cell contraction, which increases the size of the pores between the endothelial cells composing the microvasculature. The increased gap/pore size reflects in terms of the increased permeability,” [36, 79, 80, 81, 82]. If the vasodilation and increased capillary permeability have ensued, then the leaking of plasma into the interstitial spaces and also into the alveoli becomes a robust possibility. As this fluid accumulates around the alveoli, it leads to compressive pressure around the alveoli causing difficulties in their expansion during breathing and consequently, respiratory distress. The centripetal pressure around the alveoli makes it difficult for the infected person to breathe normally. The increased interstitial fluid (caused by hyperpermeability of the endothelial cells in the alveolar capillaries) seeps into the inside of the alveolar spaces, which interferes with the functioning of the surfactant [36, 79, 80, 81, 82]. The surfactant production is already diminished due to the infection and subsequent death of type-2 pneumocytes (which are primarily the surfactant producing cells). The increase of fluid accumulation could also lead to a decrease in the surfactant concentration, which may precipitate a surfactant deficiency, subsequently causing distress in breathing (increased work of breathing). The accumulation of this fluid in the alveoli and consequent drop in the surfactant concentration within the alveolar spaces is called alveolar edema. Such a condition leads to an increase in alveolar surface tension and hence respiratory distress and difficulty in breathing. An increase in surface tension leads to an increase in collapse pressure as per the Laplace's law P=2T/R where P is pressure, T is surface tension, and R is the radius of the alveoli. This mechanism indicates that such a condition is a mathematical certainty for respiratory distress resulting from alveolar collapse [36, 79, 80, 81, 82]. “This combination of factors eventually leads to a decrease in the primary function of the alveoli (the gas exchange) and consequent fall in the partial pressure of oxygen (hypoxemia; pO2). Thus, hypoxemia may present as one of the complications in the natural history of COVID-19. Apart from macrophages, the inflammatory mediators released by the demise of type-2 pneumocytes tend to bring in neutrophils which cross the alveolar membrane and start concentrating within the alveolar sac to neutralize the viruses released into the alveolar space” [36, 83, 84]. “One of the major mechanisms through which neutrophils act on viruses is to release reactive oxygen species (ROS) and proteases in certain quantities, they neutralize the virus, but in overwhelming proportions may lead to an excess ROS and proteases” [36, 83, 84]. Such a condition neutralizes the viruses and destroys the cells lining the alveoli including both type-1 and type-2 pneumocytes. As a result, the structure and, therefore, the function of the alveoli are disturbed. Since type-1 pneumocytes play an important role in gas-exchange [36, 83, 84], there is a significant drop in gas exchange. Hence, it precipitates severe hypoxemia (and a consequent drop in the partial pressure of oxygen ensues). As the cells lining the alveoli get destroyed and as the infiltration of neutrophils and macrophages as a result of inflammatory mediators released by the rupture of pneumocytes leads to increase in the cell debris within the alveolar space; the alveoli, as a result, have accumulated cell debris, degraded protein, neutrophils, macrophages [36, 37, 85, 86, 87]. “This condition referred to as consolidation severely hampers gas-exchange at the level of the alveoli again, leading to a drop in the partial pressure of oxygen and hypoxemia. Subsequently, the release of immense quantities of IL1 and IL6 seep into the bloodstream. The release of TNF-α, IL1, and IL6 comes from a vast number of alveoli within the lungs; it may lead to a significant increase of these compounds in the blood and hence travel to the central nervous system,” [36, 37, 85, 86, 87]. In response to the increased concentration of these mediators, the hypothalamus responds by releasing prostaglandins, thereby resetting the body thermostat, leading to an increase in body temperature (fever, an important presenting symptom of COVID-19). That is how the two important features (respiratory distress and fever) may be generated in the SARS-CoV-2 infection [36, 37, 85, 86, 87]. “Consolidation of cellular debris in the alveoli needs removal from the lungs, and one of the mechanisms of throwing such debris out in humans is coughing. Hence, this debris may cause cough, initially dry cough, and a productive cough. The COVID-19 patients present with this as one of the symptoms” [36, 37, 85, 86, 87]. Hence another symptom in these patients is dry and subsequently productive cough. It is worth mentioning that though certain symptoms and clinical features are frequently associated with COVID-19, the actual symptoms/clinical features may vary, and there are numerous permutations of symptoms and clinical features at the patient presentation [36, 37, 85, 86, 87]. These symptoms range from asymptomatic to cough, sneezing, sore throat, fever, pneumonia, which may, therefore, require a mechanical ventilator. In addition, lymphopenia, drop-in pO2, hepatic dysfunction, pH dysregulation, renal compromise, cardiovascular manifestations (tachycardia), GI upset, inflammatory phenotype, etc., [36, 37, 85, 86, 87]. “The low pO2 in the blood may trigger a peculiar chemo response or stimulation of chemoreceptors, which, in turn, can initiate reflux to stimulate the sympathetic nervous system (SNS) to increase the heart rate. This way, the patient might also present with an increase in pulse rate. The COVID-19 patient might also present with tachycardia. Drop-in pO2 and subsequent SNS response precipitate an increase in respiratory rate, which may also be one of the symptoms in the presenting patient. Till now, we discussed the situation when the disease is still under control” [36, 37, 85, 86, 87].
PHASE TWO: SEVERE CATEGORY
Now let us see when all these issues enter into the severe category. As more and more alveoli get involved, and an increasing number of alveoli witness consolidation, most of the inflammatory markers (IL1, IL6, and TNF-α) seep into the bloodstream, therefore, there may be an upsurge of the battery of inflammatory markers in the blood [36, 37, 85, 86, 87]. As a result, “the inflammation within the lungs (pulmonary inflammation) can lead to a systemic inflammatory response, which is a potential for septic shock. A condition that started with pneumonia may harbor the potential to culminate at the multiple systems organ failures,” [36, 37, 85, 86, 87]. Meanwhile, “Cytokine Storm arises from an exaggerated immune response to the presence of the SARS-CoV-2, with resultant overwhelming severe acute inflammation that is considered one of the most critical negative prognostic markers in COVID-19 disease” [9, 10, 11, 12].
PHASE TWO: OVERT COMPLICATED CATEGORY (DISSEMINATED INTRAVASCULAR COAGULATION)
In disseminated intravascular coagulation (DIC), the coagulation processes and fibrinolysis are dysregulated, and the result is widespread clotting with resultant bleeding. Regardless of DIC's triggering event, once initiated, the pathophysiology of DIC is similar in all conditions [88, 89, 90]. One critical mediator of DIC is the release of a transmembrane glycoprotein called tissue factor (TF). TF is present on the surface of many cell types (including endothelial cells, macrophages, and monocytes). It is not generally in contact with the general circulation but is exposed to the flow after vascular damage. For example, TF is released in response to cytokines (particularly interleukin 1), tumor necrosis factor, and endotoxin [88, 89, 90]. This process plays a significant role in the development of DIC in septic conditions. TF is also abundant in tissues of the lungs, brain, and placenta. This influence of TF helps to explain why DIC readily develops in patients with extensive trauma. Upon exposure to blood and platelets, TF binds with activated factor VIIa (usually present in trace amounts in the blood), forming the extrinsic tenase complex. This complex further activates factor IX and X to IXa and Xa, respectively, leading to the common coagulation pathway and the subsequent formation of thrombin and fibrin [88, 89, 90].
The excess activation of the coagulation cascade leads to increased circulating thrombin. Subsequently, the excess thrombin cleaves fibrinogen, which ultimately leaves behind multiple fibrin clots in the circulation. These excess clots trap platelets to become larger clots, resulting in microvascular and macrovascular thrombosis. This lodging of clots in the microcirculation, in the large vessels, and the organs leads to ischemia, impaired organ perfusion, and end-organ damage that occur with DIC [88, 89, 90]. Coagulation inhibitors are also consumed in this process. Decreased inhibitor levels will permit more clotting so that a positive feedback loop develops in which increased clotting leads to more clotting. At the same time, thrombocytopenia occurs, and this has been attributed to the entrapment and consumption of platelets. Clotting factors are consumed in the development of multiple clots, which contributes to the bleeding seen with DIC also associated with COVID-19 infection [88, 89, 90].
ASPIRIN EFFECTIVE ROLE IN COVID-19 RELATED SIRS
Aspirin is a NSAID that inhibits the systemic inflammatory response syndrome via the cyclooxygenase pathway by initially blocking prostaglandins and thromboxane synthesis. It prevents neutrophil activation and aggregation, decreases the production of superoxide radicals from activated neutrophils, and stabilizes lysosomal membranes and enzymes [20, 21, 22, 23, 24, 25, 26, 27, 28, 29]. Interestingly, by so doing, Aspirin limits the release of inflammatory mediators, and perform its role also by preventing accentuation of the pathway through the nitric oxide pathway as well described in the preceding subsections. It may thus provide hemodynamic protection and inhibit oxidant injury, especially if given early at the onset [20, 21, 22, 23, 24, 25, 26, 27, 28, 29].
ASPIRIN BENEFITS FOR THROMBO-PROPHYLAXIS IN COVID-19
As mentioned earlier, DIC and predisposition to thromboembolism are implicated in COVID-19 [91, 92]. At the cellular level, activated platelet-neutrophil interaction is essential for the formation of neutrophil extracellular traps (NETs) [93]. Through a feed-forward cycle of NETs and platelet aggregation, NETs are proposed to induce adverse cardiac and kidney events in COVID-19 patients via the production of excessive thrombosis [94]. Indeed, some studies advocate for thrombo-prophylaxis and treatment of coagulopathy in COVID-19 patients [95, 96, 97, 98]. However, these studies mostly recommend low molecular weight Heparin for the management of COVID-19 patients with coagulopathy.
Interestingly, there is evidence that Aspirin could be used for thrombo-prophylaxis in post-operative patients of joint arthroplasty [99] and prevent deep vein thrombosis in mechanically ventilated intensive care patients [100]. Besides, Aspirin has been found to help in the prevention of thrombosis through the COX-dependent and COX-independent pathways. These pathways include acetylation of prothrombin and platelet membranes and impaired formation of NETs [101]. Furthermore, compared to Heparin, Aspirin may have lesser side effects of anticoagulant-related bleeding. In light of the above, Aspirin becomes relevant as an adjuvant or monotherapy for the management of excessive thrombosis in COVID-19 patient care [99].
ASPIRIN MODULATION OF PLATELET ACTIVITY AND BIOLOGY
Aspirin inhibits platelet aggregation and prostaglandin release [17, 102]. Early studies using Aspirin radiolabeled with 14C at the acetate carbonyl carbon showed (PTGS-1/COX-1) [17, 103]. “The inhibition of platelet COX-1 by acetyl transfer is irreversible, and inhibition is maintained throughout the platelet's 10-day life. COX-1 is a bifunctional enzyme constitutively expressed in most tissues and carries out two different and sequential reactions at spatially distinct, but mechanistically coupled active sites. In normal cells, COX-1 is membrane-bound and embedded in the luminal surface of the endoplasmic reticulum and the inner as well as outer surfaces of the nuclear envelope” [17, 104, 105]. “Platelets are anucleated cell fragments and instead express COX-1 proteins in the dense tubular membrane system that originates from the demarcate membrane system during platelet biogenesis. This dense tubular membrane system plays an important role in platelet activation and is the main site for eicosanoid synthesis in platelets, as demonstrated in Figure 5” [17, 104, 105]. “The 85 kDa COX-1 homodimer contains 576 residues, and it is glycosylated at various lysine side chains. Although COX-1 is one of the few proteins that has been associated with aspirin inhibition at its active site, it is important to note that glycosylation of lysine residues enhances acetylation of other residues in this case, the serine at the active site of COX-1” [17, 106]. “Each COX-1 structural subunit incorporates three folding domains: an epidermal growth factor domain, a membrane-binding domain, and an active catalytic site. The catalytic domain contains a cyclooxygenase site that carries out the di-oxygenation of arachidonic acid to form a hydroxyl endoperoxide prostaglandin G2 (PGG2). In contrast, the adjacent peroxidase active site carries out the reduction of PGG2 to PGH2. Opposite to the membrane-active domain, within the catalytic domain, is the peroxidase active site with a heme cofactor bound to a shallow cleft” [17, 107]. “The heme group is essential to activating a tyrosyl radical in the cyclooxygenase active site for lipid peroxidation of arachidonic acid. Opposite the heme-binding peroxidase site at the top of a tunnel originating in the membrane-binding domain is the cyclooxygenase active site. Arachidonic acid binds at this site, resulting in repositioning the substrate's carboxylate for di-oxygenation,” [17, 107]. “Structural studies of ovine COX-1 treated with 2-bromoacetoxy benzoic acid suggest that the binding of arachidonic acid to the cyclooxygenase end of the active site. Consequently, the double oxygenation of arachidonic acid is inhibited by Serine 530 acetylation” [17, 107]. “Irreversible inhibition of COX-1 by Aspirin acetylation of the active site serine dramatically decreases prostaglandin biosynthesis. In platelets, COX-1 cannot be rapidly regenerated, and COX-1 activity can only be recovered by new platelet biogenesis. Synthesis of thromboxane A2, prostaglandin E2 and prostacyclin (PGI2) are the most heavily affected in aspirin-treated platelets resulting in a deficiency in the clotting mechanism” [17, 108, 109], decreased secretion of gastric mucosa, increased irritation by gastric acids [17, 110, 111, 112, 113, 114, 115, 116, 117, 118], as well as altered pathophysiological clotting, and vasodilation/constriction [17, 110, 111, 112, 113, 114, 115, 116, 117, 118]. “COX-2 is 60% identical to COX-1 at the amino acid level, and their three-dimensional structures are nearly superimposable. COX-2 is inducible, and its expression is enhanced by the same prostaglandins that are synthesized by COX-1 in platelets and epithelial cells. COX-2 is overexpressed during megakaryocytopoiesis” [17, 114] and has been identified in the cross-sectional bone marrow samples from patients with chronic myeloid leukemia and polycythemia vera [17, 116]. "Another study characterized the levels of COX-2 expression in platelets concerning COX-1, by directly measuring mRNA levels,” [17, 116]. It was found that “platelets express COX2 at levels comparable to some malignant epithelial cells, albeit at significantly lower levels than platelet COX-1. Acetylation of COX-2 in endothelial and epithelial cells inhibits PGI2 and PGE2, which have different effects on downstream processes, such as inflammation,” [17, 117, 118]. “Although aspirin-mediated inhibition of COX-1 and COX-2 results in distinct profiles of prostaglandin biosynthesis inhibition, the basis for inhibition is the blockade of prostaglandin-endoperoxide synthase and considerable reduction in multifunctional prostaglandin H2 levels. The role of COX-3 in the context of platelet biology remains unknown," [17, 117, 118]. “The COX-inhibitory activity of Aspirin is contingent on the administered dose. Low doses, those ranging from 75 to 300 mg, result in selective inhibition in platelet TXA2 production without suppressing prostacyclin (PGI2), a common platelet antagonist, and vasodilator. PGI2 is expected to be derived mainly from vascular COX-2, suggesting that COX2 inhibition is minimal in the low-dose regime [17, 117, 118]. Increased doses (>1200 mg) have analgesic and anti-inflammatory properties associated with the pathophysiological inhibition of COX-1 and COX-2. It is important to note that COX2 can also utilize arachidonic acid to synthesize lipoxins, particularly 15-hydroxyeicosatetraenoic acid (15-HETE). This biosynthetic route is expected to remain intact even after COX-2 acetylation” [17, 117, 118]. “This differential inhibition of COX activities can be explained, in part, by the relative inhibitory potency of Aspirin. Although Aspirin is typically thought of as a non-specific COX inhibitor, it is highly selective for COX-1 versus COX-2. As seen in Blanco et al.,” [17, 119], the IC50 of Aspirin for COX-1 is approximately 3.5 μM, while the IC50 for COX-2 is approximately 30 μM. While the Aspirin reactive active sites of both enzymes are homologous, acetylation of Ser-516 of COX-2 results in only partial inhibition of catalytic activity [17, 120, 121]. “Given the achievable serum concentration in the low-dose regime (~7 μM), it is unlikely that the COX-2 is more than 5% acetylated while platelet-derived COX-1 is likely to be >70% acetylated” [17, 121]. “This process suggests that regular low-dose Aspirin will invariably maintain COX-1 inhibition in circulating platelets, with minimal effect in the inhibition of peripheral COX-2.
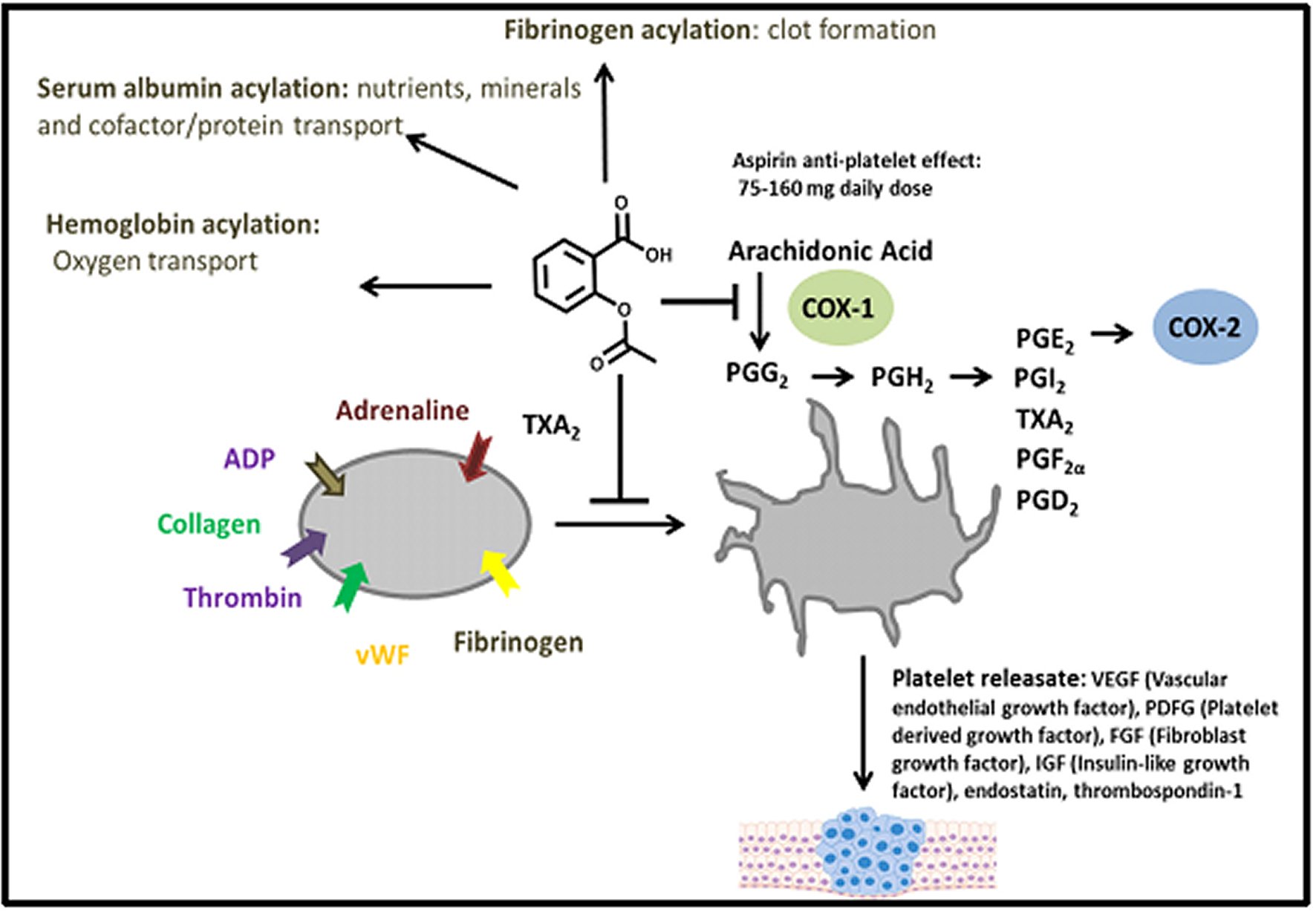
Figure 5. Activation of platelet Platelet activation is initiated by multiple stimuli including thrombin, ADP-Adenosine-diPhosphate, and fibrinogen.TXA2: Thromboxane A2; COX: Cyclooxygenase. PGG2: Prostaglandin G2; PGH2: Prostaglandin H2; PGE2: Prostaglandin E2; PGI2: Prostaglandin I2; PGF2α: Prostaglandin F2α; PGD2: Prostaglandin D2; vWF: von Willebrand factor.
Similarly, Block et al., in 2012, in their study on Aspirin's effectiveness in reducing cardiovascular disease events, is inadequate in some individuals, a phenomenon termed aspirin ‘resistance.' They submitted that “a potentially beneficial effect on platelet function occurred within four hours after the ingestion of low-dose Aspirin and eicosapentaenoic acid and docosahexaenoic acid (EPA+DHA) in healthy adults” [122].
ASPIRIN MODULATION OF PLATELET ACTIVITY AND BIOLOGY
Bosetti C et al. conducted a systematic review and meta-analysis of all observational studies on aspirin and cancers of the digestive tract sites published through March 2019 to provide an up-to-date quantification of this association [123]. They submitted that for colorectal cancer, and aspirin dose between 75 and 100 mg/day conveys a risk reduction of 10% and a treatment of 325 mg/day of 35%. For all neoplasms, except head and neck cancer, inverse duration-risk relations with aspirin use are found. Interestingly, the present comprehensive metaanalysis supports and further quantifies the inverse association between regular aspirin use and the risk of colorectal and other digestive tract cancers, including some rare ones. The favorable effect of aspirin increases with longer duration of use, and, for colorectal cancer, with increasing dose [123].
Furthermore, “Ishikawa et al. analyzed 51 randomized controlled trials (RCTs), and the cumulative evidence strongly supports the hypothesis that daily use of aspirin results in the prevention of cardiovascular disease (CVD), as well as a reduction in cancer-associated mortality” [17, 124]. “Six other trials also showed reduced gastrointestinal cancer incidence after three years of aspirin use (OR, 0.76, 95% CI, 0.66–0.88; p = 0.0003) with statistically significant reductions also observed for esophagus, colon, and lung cancers. All cancer-preventive, incidence, mortality, and metastasis effects were more pronounced with increased duration of the scheduled trial treatment. Importantly, all fatalities from major extra-cranial bleeds were lower in aspirin than controls, with a favorable risk/benefit ratio. Other studies have also demonstrated reductions in cancer incidence” [17, 125], particularly for cancers of the gastrointestinal tract, along with cancer-related [126, 127, 128] and all-cause mortality [17, 126, 127, 129]. “Other evidence that NSAIDs and aspirin prevent cancer was shown in multiple clinical trials involving short-term aspirin use at varying doses that reduced colorectal adenomas. For patients with a previous history of colorectal cancer (CRC) [17, 130] or colorectal adenomas”, [17, 131, 132], those taking aspirin showed fewer new adenomas compared to no-aspirin controls. In The Colorectal Adenoma/Carcinoma Prevention Programme (CAPP) trial, the long-term use of aspirin in a cohort with hereditary CRC (Lynch Syndrome) patients revealed an HR of 0.63 (95% CI, 0.35–1.13; p=0.12) [17, 133]. Looking forward, “in the ASPirin Intervention for the REDuction of colorectal cancer risk (ASPIRED) trial, the effects of aspirin will be examined using various biomarker endpoints” [17, 134]. “ASPIRED is designed as a prospective, double-blind, multidose, placebo-controlled, biomarker clinical trial of aspirin use in a cohort of patients previously diagnosed with colorectal adenoma. Subjects (n = 180) will be randomized to low-dose (81 mg/day) or standard-dose (325 mg/day) aspirin or placebo. These individuals will give lifestyle, dietary, and biometric data. They will also provide urine, saliva, blood specimens, stool, grossly normal colorectal mucosal biopsies, and cytology brushings collected during flexible sigmoidoscopy,” [17, 134]. “As biomarker endpoints, the effect of aspirin on urinary prostaglandin metabolites (PGEM; primary endpoint), plasma inflammatory markers (macrophage inhibitory cytokine-1 (MIC-1)), colonic expression of transcription factor binding (transcription factor 7-like 2 (TCF7L2)), colonocyte gene expression, including hydroxy prostaglandin dehydrogenase 15-(NAD) (HPGD) as well as Wnt-signaling transcripts, colonic cellular nanocytology, and oral and gut microbial composition and function will be assessed” [17, 134]. This study will help further to understand the impact of aspirin on CRC adenoma biology, and may provide insight into the role of platelets in the chemopreventive mechanisms of aspirin [17, 134].
Surprisingly, most Africans are eager to see the desired transformation in our public health systems. Unfortunately, the political will to invest in public health infrastructure is lacking [135]. Also, the system is characterized by human resources shortage and diverted resources, which significantly impacted the provision for emerging COVID-19 pandemic –related care [135]. Interestingly the monumental breakthroughs in research development for bio-therapeutics and vaccines in African countries appear a mirage even with extensive past study experience with such products from China and the Western world [135]. Finally, notwithstanding these challenges in our public health systems as elaborated, the facts are that enormous capacities exist that can be harnessed in African countries for the COVID-19 preparedness and response [135].
The overall key panel message from this presentation is shown in Table 1.
RECOMMENDATIONS
There is a critical need to conduct a comprehensive systematic review and meta-analysis of all observational studies on Aspirin as effective anti-inflammation, analgesic, and antipyretic, as well as a potentially beneficial effect on platelet function, published till 2020 to provide an up-to-date quantification of this association.
Most wanted is The RECOVERY II (Randomized Evaluation of COVID-19 Therapy) trial to be established as a randomized clinical trial to test a range of potential treatments for COVID-19, including low-dose Aspirin as an anti-inflammatory and antithrombotic treatment.
A Trial Center and Team are to be established to serve as a coordinating unit for The RECOVERY II (Randomized Evaluation of COVID-19 Therapy) trial.
All other operational modalities and acquisition of grants are to be worked out by The RECOVERY II Trial Research Coordinating Team.
There is advocacy for stringent adherence to the declaration of Helsinki in the conduct of clinical trials.
The need to establish a worldwide Pandemic research think-tank to Debate, Review, and Evaluate and formally disclose significant clinical research findings before going to the print media.
There is a call for more responsible leadership and the use of guided utterances in matters regarding the distribution and use of pharmaceutical agents to forestall catastrophes.
There is also advocacy for stringent regulations regarding Pharmaceutical agents, its sale, distribution, and consequent use.
CONCLUSIONS AND FUTURE DIRECTIONS
At this juncture, the world patiently awaits announcement by scientists the development of a breakthrough after years of painstaking research, for treatment that costs pennies a pill, saves lives, and could reduce healthcare spending by nearly $700 billion in the coming years. Besides, you wouldn't even need a prescription to get it and in the context of combating severe complications following COVID-19 infection. Perhaps this all sounds too good to be accurate, but, according to a new study, wonder drug is already here. It's called Aspirin. Interestingly Aspirin has been associated with a reduced risk of colorectal cancer, and possibly a few other digestive tract cancers. Current evidence further quantifies the inverse association between regular aspirin use and the risk of colorectal and other digestive tract cancers, including some rare ones. The favorable effect of Aspirin increases with longer duration of use, and, for colorectal cancer, with increasing dose.
Aspirin-mediated acetylation of cyclooxygenase enzymes is believed to be the primary mechanism for anti-inflammatory, antipyretic, and antiplatelet effects. While the platelet dependent effects of Aspirin have been extensively studied in the context of COX-1, the role of non-canonical (e.g., nonCOX) aspirin-mediated acetylation in platelets remains largely unknown. Numerous proteomic studies have attempted to identify the “off-target” effects of Aspirin in both standard human and cancer cell lines, and such studies have proved valuable in understanding prophylactic aspirin effects.
As our understanding of the cross-talk between platelets, tumor cells, and the immune system expands, the COX-dependent and COX-independent effects of Aspirin in modulating interactions at the biochemical level can now be directly addressed.
The emerging interest is to conduct further study on these principal mechanisms by which Aspirin inhibits acute inflammation and alters platelet-biology. Therefore, it provides evidence for Aspirin as a novel therapeutic drug for combating severe acute inflammation and thrombosis associated with the cytokine storm in COVID -19 patients. Most wanted is The RECOVERY II (Randomized Evaluation of COVID-19 Therapy) trial to be established as a randomized clinical trial to test a range of potential treatments for COVID-19, including low-dose Aspirin as an anti-inflammatory and antithrombotic treatment.

