










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkArchivos Españoles de Urología (Ed. impresa)
versão impressa ISSN 0004-0614
Arch. Esp. Urol. vol.61 no.2 Mar. 2008
MONOGRÁFICO: REFLUJO VESICOURETERAL
Embriología y genética del reflujo vesicoureteral primario y de la displasia renal asociada
Embryology and genetics of primary vesicoureteral reflux and associated renal dysplasia
Antonio Morales Martínez, Rocío Calvo Medina, Mercedes Chaffanel Peláez, Alberto Bueno Fernández, Carlos Miguelez Lago y Emilia del Castillo Acedo del Olmo.
Servicio de Urología, Servicio de Nefrología y Servicio de Enfermedades Genéticas. Hospital Materno Infantil. Hospital Universitario Carlos Haya. Málaga. España.
Dirección para correspondencia
RESUMEN
Objetivo: Realizar una aproximación a la embriología y genética del reflujo vesicoureteral (RVU) y de la nefropatía de reflujo (NR). Reconocer los patrones de asociación familiar del RVU y tratar de ver en qué casos se deberían considerar estudios genéticos a estos pacientes y, en tal caso, qué tipo de estudios. Por último, estudiar qué tipo de asociación presentan estos dos fenómenos (RVU y NR).
Métodos: Revisión bibliográfica de artículos relacionados hasta junio de 2007.
Resultados: Se reconocen dos tipos de RVU primario según la presentación: aislado y sindrómico; de este último se conocen sus mecanismos de transmisión y sigue patrones de herencia mendeliana. Los estudios epidemiológicos ponen de manifiesto que el RVU aislado también presenta asociación familiar, y es objeto de estudio el patrón de herencia del mismo; la mayoría de los autores sostienen la idea de que se trata de un fenómeno genéticamente heterogéneo en el que interactúan diferentes genes y efectos medioambientales. Son múltiples los genes candidatos implicados. Las características del RVU (expresividad y penetrancia variables, resolución espontánea) hacen difícil seleccionar aquellos pacientes subsidiarios de estudio genético. A pesar del tratamiento del RVU, la incidencia de fallo renal crónico secundario al mismo no ha descendido. Algunos de los genes candidatos en estudio para el RVU se han identificado como reguladores de la embriogénesis de la yema ureteral, paso clave para el desarrollo del riñón y tracto urinario. Análisis en ratones y humanos sugieren que parte del daño renal asociado al RVU es congénito y debido a una malformación congénita; por lo que la asociación entre RVU y fallo renal puede deberse a un defecto genético del desarrollo y no a la mala evolución del paciente con RVU. La investigación en animales es un paso clave para profundizar en los conocimientos en este tema (genes candidatos y asociación de RVU con NR).
Conclusiones: Es importante conocer los patrones de asociación familiar del RVU (aislado y sindrómico) para dar consejo genético cuando sea posible. Por ello, en todo paciente con RVU, son necesarias una anamnesis y exploración específicas y dirigidas. La indicación actual de estudio genético tiene limitaciones. Se realizará diagnóstico prenatal si existe RVU sindrómico con mutación conocida, expresividad fija y penetrancia completa o si las manifestaciones conllevan riesgo vital. Los datos epidemiológicos y estudios de laboratorio nos orientan a la posibilidad de nefropatía congénita asociada en casos de RVU grave.
Palabras clave: Reflujo vesicoureteral. Nefropatía de reflujo. Genética del RVU. Desarrollo renal y tracto urinario. Malformación renal. Embriología.
SUMMARY
Objectives: The main reasons of this review are: To determine some of the embryological and genetic mechanisms of vesicoureteral reflux (VUR) and associated congenital reflux nephropathy (NR); recognize different patterns of familiar clustering and identify appropriate cases where genetic counselling and investigations might be indicated; and finally, to establish the association of these phenomena (VUR and NR).
Methods: Bibliographic search of related articles until June 2007.
Results: There are two kinds of primary VUR: isolated VUR and syndromic VUR; the last one has an inherited Mendelian transmission and we know the mechanisms. Epidemiological studies seem to demonstrate that isolated VUR also presents familiar clustering and its inheritance pattern is the main object of interest in some studies; most authors support the hypothesis that VUR is genetically heterogeneous and is caused by a number of different genes acting with random environmental effects. There are lots of candidate implicated genes. The characteristics of VUR (incomplete penetrance, variability of expression, spontaneous resolution
) make difficult to configure a selection of patients subsidiary of genetic study. Despite different treatment options, the incidence of renal chronic failure secondary to VUR has not decreased. Some of the candidate genes identified regulate the position of ureteral budding, a critical step in both kidney and urinary tract development. Analysis of data from humans and mice suggests that some of the renal damage associated with VUR is congenital and is due to a kidney malformation. Therefore, in these cases, the association of VUR and renal failure may be caused by a genetic defect affecting the formation of the kidney and the urinary tract and not by evolution of VUR. Investigation in animals is fundamental to know more about this issue (candidate genes and VUR-NR association).
Conclusion: It is important to learn patterns of familiar clustering of isolated and syndromic VUR to offer genetic counselling if possible. For this reason, we should be screening carefully all patients suffering from VUR. It is known that limitations in actual indications of genetic study exist. Prenatal diagnosis may be realized if there is a syndromic VUR with known mutation, invariable expressivity or if clinical manifestations involve risk of death. Epidemiological data and laboratory studies may give us guidance to elicit new cases of nephropathy associated to severe VUR.
Key words: Vesicoureteric reflux. Reflux nephropathy. Genetics of VUR. Kidney and urinary tract development. Kidney malformation.
Introducción
ASOCIACIÓN FAMILIAR DEL RVU
El Reflujo vesicoureteral (RVU) primario es un defecto congénito del tracto urinario que consiste en el paso retrógrado de orina de la vejiga al riñón, descartando alteraciones neuromusculares u obstructivas que lo expliquen (1). Se estima que su prevalencia en la edad pediátrica está en torno al 1% de todos los niños (2,3), aunque dada la variabilidad de las manifestaciones clínicas, y las distintas características de los medios diagnósticos, ésta puede estar infravalorada.
Los datos recogidos en las series americanas y europeas informan de que el 8% de los pacientes con RVU desarrollan fallo renal crónico como consecuencia de la denominada Nefropatía por Reflujo (NR), (4,5) representando ésta el 25% de las causas de enfermedad renal crónica (1,2). Dado que en ocasiones el RVU no tiene manifestaciones clínicas, probablemente exista un porcentaje de pacientes con RVU no diagnosticado, asociado a diferentes grados de hipoplasia y/o displasia renal (RHD). Ésta es, a su vez, la enfermedad subyacente en más de un tercio de los niños con enfermedad renal crónica (6,7).
En nuestra experiencia diaria, podemos sospechar que el fenómeno del RVU presenta un componente familiar, refrendado en numerosos estudios actuales. Así, se han registrado diferentes incidencias de RVU aislado, en análisis de poblaciones con diferencias étnicas (8); tasas de concordancia de RVU del 80-100% en gemelos monocigóticos (frente al 35-50% en los dicigóticos.) (9); incluso otros autores refieren que el 30-50% de los hermanos de afectos de esta enfermedad, presentan RVU no diagnosticado, definiendo así claramente el agrupamiento familiar de esta patología (10).
Por todo esto, el componente genético de este fenómeno ha sido estudiado en profundidad, siendo objeto en los últimos años de muchas publicaciones en revistas especializadas. De este modo se han sugerido varios patrones que intentan explicar el comportamiento genético del RVU [autosómico dominante (AD) con penetrancia incompleta (3,11), autosómico recesivo (AR) (12), ligado al sexo (13), de herencia multifactorial (2, 14,15)] pero tan sólo Chapman (3) realiza un análisis de segregación complejo que determina que el patrón que mejor puede explicar las asociaciones epidemiológicas y familiares de RVU es el de un gen dominante simple. Robert Mak (1) habla de este último análisis presumiendo que se trataría de una mutación, en la que el alelo mutado domina sobre el no mutado y que la frecuencia de presentación sería de 1/600 (hipótesis que haría de esta enfermedad la más común en humanos transmitida con herencia dominante).
Es indudable es que existen anomalías nefro-urológicas congénitas derivadas de alteraciones en el complejo desarrollo embriológico del sistema urinario y renal, así como una serie de enfermedades multisistémicas de reconocida etiología genética, en las que algunas de estas anomalías están presentes, entre ellas el RVU (16). Las anomalías nefrourológicas comentadas se han denominado clásicamente por su acrónimo en inglés CAKUT (congenital anomalies of the kidney and urinary tract).
Por otro lado, cabe señalar que otros estudios descriptivos muestran que los miembros de familias en las que existe RVU y NR van a tener una mayor posibilidad de presentar NR, que los de aquellas familias que presenten solamente RVU (17-19). Igualmente señalan que los miembros de familias con pacientes afectos de RVU grave, van a tener mayor probabilidad de presentar NR al nacimiento (20).
Desarrollo embriológico
En este punto es interesante conocer la relación que se establece entre el desarrollo del RVU y la posibilidad de aparición de un riñón hipoplásico/displásico asociado. Esta teoría propuesta por varios autores, sostiene que la nefropatía que se asocia al RVU se debe a un mecanismo común que afecta al mismo tiempo en el desarrollo embriológico del sistema urinario y renal (21,22).
Para entender la asociación de RVU y malformaciones renales es preciso comprender el desarrollo del tracto urinario y del riñón. En los vertebrados, el riñón procede de los conductos mesonéfricos, una pareja de cordones epiteliales que se extienden a lo largo del eje antero-posterior del embrión (23). La yema ureteral (YU) se extiende desde el conducto mesonéfrico subiendo hasta el blastema renal. En la 4ª semana del desarrollo embrionario, señales recíprocas entre la YU y el blastema metanéfrico adyacente, inducen que el extremo distal de la YU comience a bifurcarse y a formar los conductos colectores. Al mismo tiempo el blastema metanéfrico se epiteliza formando las nefronas. El extremo proximal de la YU permanece unido al conducto mesonéfrico y formará el uréter distal. El conducto néfrico común, que es el segmento del conducto mesonéfrico que enlaza el uréter y el seno urogenital (primitiva vejiga), está programado para la muerte celular, lo cual permite al uréter insertarse dentro de la vejiga (trayecto submucoso) y desarrollar una capa muscular que conecta con el trígono vesical (Figuras 1 y 2).
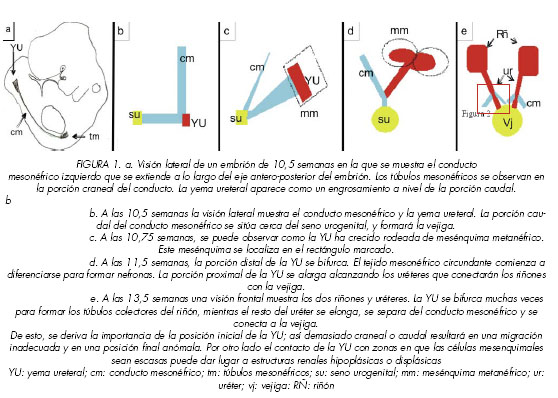

Numerosos estudios hablan de que la posición de la YU en el conducto mesonéfrico es fundamental para el desarrollo del riñón y el tracto urinario (24, 25). De este modo una YU situada demasiado craneal resultaría en un orificio situado en la uretra o en estructuras genitales; mientras que la posición inicial de la YU demasiado caudal llevaría a una inserción en el trígono demasiado craneal y como consecuencia una unión ureterovesical corta o lateral al trígono. Mackie y Stephens (26) examinaron lactantes postmortem afectos de duplicidad renal y hallaron que la posición del orificio ureteral se correlacionaba con la morfología renal de tal modo que resultaba un riñón displásico en caso de posición del orificio lateral al trígono o en la uretra.
Por otro lado Murer (27) apunta que la interacción necesaria entre la YU y el blastema metanéfrico para el crecimiento y diferenciación, también se altera dependiendo de la posición de la YU, ocurriendo que en casos de contacto con zonas en que las células mesenquimales son escasas, se desarrollarán riñones hipoplásicos o displásicos. De acuerdo a estas teorías la hipoplasia/displasia renal no siempre sería consecuencia del RVU crónico y sus manifestaciones, sino de la unión aberrante de la YU con el mesénquima metanéfrico.
Por este motivo es de gran importancia el estudio de los factores que determinan la posición de la YU entre los que se encuentran moléculas de señalización y factores de transcripción expresados en el conducto mesonéfrico y en el blastema metanéfrico (Tabla I), de tal modo que alteraciones en la expresión de estas moléculas suponen una alteración del desarrollo que conllevará tanto a malformaciones renales como del tracto urinario (28).

Bases genéticas del RVU
A la hora de hablar de transmisión genética del RVU y concreción de las estructuras implicadas el espectro de trabajo es muy amplio puesto que, como ya suponemos, se trata de encontrar aquellos genes que pueden afectar al proceso de desarrollo del tracto urinario y renal, hecho que se ha comprobado en la experiencia de laboratorio con ratones. Así, al hablar de genes que alteran el desarrollo, predisponentes para la aparición de RVU tenemos que hablar de:
1º Mutaciones en genes que se han demostrado en pacientes con hipo-displasia renal sindrómica que siguen patrones de herencia mendeliana (estos genes han demostrado estar implicados en el RVU),
2º Genes concretos en los que diferentes alteraciones (delecciones, traslocaciones ) pueden dar lugar a alteraciones nefro-urológicas;
3º en último lugar no se debe olvidar que este tema es terreno de investigación en la actualidad y que la experiencia en animales es vital para su conocimiento más completo.
I.- SÍNDROMES DE HERENCIA MENDELIANA CON HIPO/DISPLASIA RENAL
Aunque la mayoría de los pacientes con RVU no tienen otras malformaciones orgánicas, mutaciones en genes específicos han sido identificados en pacientes con RVU como parte de un síndrome más complejo (Tabla II), algunos de los cuales son los siguientes (27):

- Síndrome coloboma renal (RCS). Alteraciones del tracto urinario, oculares y óticas. Experiencias en ratones y humanos. La mayoría de las alteraciones se dan en el exón 2. Presenta gran variabilidad fenotípica incluso intrafamiliar; se ha visto en alteraciones aisladas como hipoplasia renal o RVU aislado. AD. Locus 10q24–25.
- Síndrome branquio-oto-renal (BOR). Alteraciones del tracto urinario, auriculares y quiste branquial. AD con penetrancia incompleta y expresividad variable. Locus 8q13.3.
- Síndrome Townes-Brocks. Presenta sordera, dermopatía, ano imperforado, trifalange del pulgar y alteraciones renoureterales. AD, se han reconocido 35 mutaciones diferentes. Cromosoma 16q12.1.
- Síndrome hipoparatiroidismo-sordera neurosensorial-enfermedad renal. GATA 3. AD. Locus 10p14-15.
(El sitio web Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db_OMIM) presenta una lista de más de 100 síndromes CAKUT reconocidos, muchos de los cuales han sido definidos genéticamente)
En este punto es muy interesante un estudio reciente multicéntrico (29) . En él de un total de 466 niños con estadío 2 a 4 de enfermedad renal crónica, definida por Filtrado Glomerular (GFR) alterado (FGR=15 a 75 ml/min por 1`73 m2) se seleccionan 175 pacientes diagnosticados por ecografía de tamaño renal pequeño (en longitud o volumen) o con pérdida de diferenciación córtico- medular. De ellos se excluyen aquellos que presentaban de forma asociada anomalías como válvula uretral posterior o malformaciones vesicales primarias, así como aquellos en que existían síndromes complejos que afectaban al riñón, diferentes a los estudiados en este ensayo. De este modo quedan 100 pacientes en 99 familias diferentes (2 eran gemelos) en los que el 12% presentaban historia familiar positiva y el 23% anomalías extrarrenales. En este grupo se estudian con amplificación por Reacción en Cadena de la Polimerasa (PCR) las secuencias genómicas de DNA para TCF2, PAX2, EYA1, SIX1 y SALL1 así como cribaje para delección de TCF2 y EYA1 resultando que variantes secuenciales en dichos genes de desarrollo renal fueron encontrados en 18 pacientes, la mayoría de ellas en TCF2 y PAX2.
Lo realmente importante de este ensayo es ver qué manifestaciones clínicas tanto renales como extrarrenales acompañan a la mutación encontrada. Sólo en cinco de los dieciocho niños (5/18) se había sospechado previamente por la clínica el síndrome asociado al gen mutado hallado; de acuerdo con esto, uno de cada seis niños que presentan RHD se espera que pueda tener alteraciones en uno de los cinco genes estudiados aquí.
Es interesante la diversidad de las manifestaciones; así, por ejemplo el espectro clínico de los pacientes con mutación del gen PAX2 incluye hipoplasia renal, displasia no quística o quística (glomeruloquística o multiquística) reafirmando la hipótesis de que alteraciones en el gen PAX2 llevan a una obstrucción ureteral temprana durante la nefrogénesis con las consecuencias de los fenotipos citados. También se comprobó que en la mayoría de los pacientes con anomalías oculares asociadas, éstas fueron descubiertas en la reevaluación de los mismos por no haber dado manifestaciones anteriormente.
Otro punto de interés es el hecho de que seis de los ocho niños afectos de alteraciones en TCF2 presentaban lesiones quísticas. Visto de otra manera el 22% de todos los niños del ensayo con RHD quística presentaban mutaciones o delecciones en el gen TCF2. De estas ocho familias se extrajeron muestras en cuatro de ellas viendo que existía mutación en dos de las mismas (asumiendo que eran de novo las otras dos) y comprobando antecedentes familiares de diabetes en tres de las ocho.
Una lectura también de relevancia es que de los pacientes afectos de BOR, aquellos que presentaron la variante que afecta a SIX1 demostraron una rápida evolución a insuficiencia renal crónica y a displasia del disco óptico.
Todo esto nos señala la importancia de emparejar alteraciones genéticas con manifestaciones clínicas para vislumbrar si las primeras nos van a resultar de utilidad en la práctica clínica.
II. GENES CANDIDATOS
Están en investigación múltiples genes candidatos a ser agentes influyentes en el desarrollo del tracto urinario y renal y, por tanto, en la posible aparición de RVU.
Éstos se han importado desde la experiencia clínica, desde las asociaciones del RVU con otra patología (por ejemplo dada la supuesta asociación de RVU con infecciones de orina se ha propuesto que en el cromosoma 6 del sistema de antígenos de histocompatibilidad debe haber un locus que se asocie a la presencia de RVU, incluso a la evolución hacia NR (30-33); y, también de la investigación. En la Tabla III se adjuntan los genes más estudiados a este respecto con las manifestaciones renales que se derivan de ellos así como las referencias de los investigadores que han estudiado dichas asociaciones. Algunos de ellos son los siguientes:

- UPKIII una proteína de membrana que juega un papel fundamental en la estructura y función urotelial y, por tanto, parece importante en la aparición de RVU. Además, dado que forma parte de dicha estructura el contenido en proteínas solubles excretadas en orina puede variar, pudiendo esto ser objeto de estudio y se comentará más abajo (3, 34,35).
- ROBO2 receptor axonal en el blastema metanéfrico de una glicoproteína (Slit2) secretada en la YU (34,35).
- Enzima Convertidor de la Angiotensina (ACE) y el receptor Agtr2 parecen tener un papel fundamental en guiar la YU a su ubicación adecuada sin embargo la mutación de éste solamente no parece dar lugar a la aparición de RVU, por lo que parece más bien un gen necesario, modificador para el desarrollo del fenotipo (34-36)
Los estudios que tratan de dilucidar el comportamiento genético y los posibles locus asociados son escasos, ya que son de gran dificultad técnica. Destaca el realizado por Feather (37) en 7 familias europeas; se trata de la primera búsqueda genómica en RVU no sindrómico y NR en el que además de analizar asociaciones de aparición de RVU con diferentes locus en el mapa genético apunta que las conclusiones de Chapman parecen confirmarse en su estudio. Sin embargo concluye que la herencia dominante del RVU debe darse tan solo en una pequeña proporción de la población mientras que en la mayor parte de la misma la herencia es poligénica.
Todos estos datos apoyan la hipótesis de que el RVU es genéticamente heterogéneo causado por una diversidad de genes diferentes actuando solos o en combinación. (2,27,28,37).
Una importante aplicación, derivada del estudio genético en humanos, es la posibilidad de realizar un análisis específico de la orina, denominado análisis proteómico que comentaremos a continuación.
III. IMPORTANCIA DE LOS MODELOS EN RATONES
Hay una gran cantidad de ensayos y estudios realizado en animales. Los más destacables e importantes se han hecho en ratones habiéndose diseñado mapas de alta resolución genómicos disponibles y análogos entre las dos especies.
De este modo los modelos en ratones (28) para las mutaciones Pax21 Neu+/- (38) y Hoxb7/Ret+/- llegan a la misma propuesta de Mackie y Stephens que postulan que una YU caudalmente situada conducirá a un riñón y uréter malformado. Murawski (28) especula que una YU anormalmente situada en un mesénquima con escasa capacidad para responder a señales de inducción llevará a un orificio del uréter situado demasiado lateral y a un uréter intravesical acortado debido a que el contacto es demasiado temprano y la migración inadecuada. De acuerdo con esto, estos autores (en estudios aún no publicados) han comprobado que el tamaño renal y el tamaño del uréter intravesical presentan una asociación estadísticamente significativa concluyendo que están correlacionados.
Una de las experiencias animales con una posible mayor aplicación en la clínica es el desarrollo de anomalías urológicas (RVU, hidronefrosis) y función renal alterada en ratones con disrupción del gen de la uroplakina III (1) que conducen a un urotelio alterado. Parece que este urotelio alterado también puede desempeñar un papel en el desarrollo de RVU en humanos. Se sabe que el urotelio secreta proteínas en la orina (39,40) por lo que diferentes patrones de proteínas en orina pueden traducir una mutación genética causante o predisponerte de RVU primario (con lo que estas proteínas podrían ser marcadores de este fenómeno.) Los estudios realizados en este punto por Mak y cols. (41) se basan actualmente en la secuenciación de las moléculas en muestras de orina obtenidas en pacientes afectos de NR, este es el llamado Análisis Proteómico de la orina. Concluyen que los futuros trabajos en este campo deben basarse en la identificación y secuenciación de estas moléculas.
La conclusión de la experiencia animal es que mientras no hay genes específicos humanos ligados al RVU sí que los hay en modelos de ratones de tal modo que aquellos identificados habría que trasladarlos al mapa genómico humano y comprobarlos.
Discusión
Dado el conocimiento cada vez más amplio de las bases moleculares y genéticas del RVU familiar se nos plantea la posibilidad de diagnóstico prenatal, sin embargo, en este punto hay que ser muy cautos porque como ya se ha visto existen casos en que la misma mutación se expresa con manifestaciones más o menos severas y no disponemos de factores pronósticos para predecirlas (42).
Sin embargo, otros autores reconocidos (29) son más categóricos y optan por el consejo genético a todos aquellos pacientes afectos de síndromes con RHD y herencia mendeliana AD, aunque estos tengan una expresividad variable y en ocasiones penetrancia incompleta. También se aconseja ampliar el estudio genético a familiares de pacientes afectos pues, como demuestra Weber en su serie, la más representativa hasta el momento, el porcentaje de portadores de la alteración en las familias (en las que se pudo analizar) era del 50 y 66 % respectivamente en los genes implicados más frecuentemente (TCF2 y PAX2), porcentaje que se incrementa si incluimos historia familiar positiva clínica.
También es interesante el estudio genético de aquellos pacientes afectos de RHD ya que, según hemos visto, estos pacientes pueden tener alteraciones genéticas lo cual nos ayudará a descubrir las comorbilidades de dichas enfermedades genéticas. Dichos autores recomiendan el estudio de alteraciones no sólo en aquellos pacientes con historia familiar presente, sino en todos por la posibilidad de que se trate de mutaciones de novo. Las determinaciones a realizar varían según el autor.
De forma más específica, los pacientes afectos de RVU ¿serían candidatos a estudio genético? ¿y sus familiares?
Posiblemente de forma análoga a lo anteriormente descrito sería de utilidad completar el estudio por parte del urólogo realizando un perfil genético a aquellos pacientes afectos de RVU con características asociadas, así sería en pacientes con:
RHD o fallo renal crónico no explicado por otra causa,
Alteraciones extrarrenales que puedan tener algún significado sindrómico
y quizá en aquellos con historia familiar de RVU y/o de NR.
Los signos y síntomas extrarrenales tienen una doble vertiente desde el punto de vista de la práctica clínica: por un lado, el hecho de estar presentes nos refuerzan la necesidad de estudio genético; por otro lado un estudio genético positivo nos señalará la necesidad de una exploración minuciosa e, incluso, de derivación al especialista (fondo de ojo, metabolopatía, alteraciones endocrinológicas, audiometría ).
En la Tabla IV se adjuntan datos que se han de recoger en la anamnesis y exploración física de todo paciente con RVU que, en caso de resultar positivos, nos reforzarían la idea de solicitar estudio genético o consulta con el especialista.

Posiblemente, al menos hasta la fecha, no sería de interés en aquellos pacientes afectos de RVU primario aislado en que no se encuentran ninguna de las características anteriormente mencionados ya que, hasta el momento, no hay una clara asociación de RVU aislado con genes concretos.
El conocimiento cada vez más amplio de las bases genéticas y moleculares del RVU promete la posibilidad de tests no invasivos para su diagnóstico y probablemente proveerá pruebas de screening directos. Sin embargo, dado que el RVU primario aislado parece comportarse como un desorden poligénico, los diseños de dichos tests probablemente llevarán tiempo y está por ver si tendrán aplicación práctica.
Posiblemente el avance en investigación en este sentido nos permitirá reconocer la utilidad de estos análisis en familiares de los pacientes así como la posibilidad de consejo genético. Sin embargo en el caso del RVU la dificultad de los estudios es manifiesta dadas sus características: herencia multifactorial, variabilidad de manifestaciones y resolución espontánea en muchos casos.
En la Figura 3 se muestra una propuesta sobre qué tipo de medidas se deben tomar en el momento actual en el campo de la genética según el patrón de presentación del RVU en un paciente.

A) En el caso de RVU aislado hoy en día no cabe otra actuación que el consejo genético advirtiendo que la tasa de transmisión varía entre el 4-7% al 50% según consideremos que se trata de herencia poligénica o de herencia AD respectivamente y en estos últimos no se debe olvidar que no siempre la penetrancia es completa así como la variabilidad de las manifestaciones.
B) En los casos de RVU sindrómico la tasa de afectación en padres y en futura descendencia se rige por los patrones clásicos de la herencia (Figura 3) asumiendo que se puede hacer estudio molecular en caso de que exista mutación conocida.
A pesar de los múltiples tratamientos medico-quirúrgicos para tratar el RVU (43-45), la denominada NR sigue siendo la tercera y quinta causa más frecuente de fallo renal en las bases de datos pediátricas y de adultos respectivamente (28). Por otro lado la teoría clásica que propone que el daño en el parénquima renal en los pacientes afectos de RVU (es decir la NR) es debido a las infecciones de repetición en estos pacientes pierde paulatinamente fuerza ya que se ha demostrado en numerosos estudios que estos pacientes ni tienen mayor cantidad de reinfecciones, ni siquiera una mayor tasa de pielonefritis (45-47). En este punto es importante señalar que tanto series nacionales (48) como internacionales (30,49,19,20) recogen el carácter congénito de la nefropatía en aquellos pacientes afectos de RVU grave que, en la mayoría de los casos, es bilateral.
Por otro lado, evidencias de humanos afectados sugieren que tanto el RVU como la NR parecen resultar de una anomalía en la posición de la yema ureteral a lo largo del conducto mesonéfrico. Las experiencias en ratones demuestran que cuando hay alteraciones en los genes que regulan la posición de la YU se ponen de manifiesto tanto el RVU como la NR al nacimiento.
De acuerdo a estos datos epidemiológicos y a la experiencia de laboratorio parece que se puede concluir que tanto la NR congénita asociada a RVU (al menos en sus grados más graves) como el propio RVU son dos manifestaciones de un mismo problema en el desarrollo embriológico del sistema renal.
Es quizá en este punto en el que se podría actuar; de ahí la importancia que han adquirido los modelos animales para la búsqueda de genes que conducen al RVU. Los avances en este terreno ofrecen posibilidades muy optimistas acerca del manejo de estos pacientes:
Predicción de qué pacientes tienen riesgo de desarrollar NR.
Posibilidad de sustituir el screening cistográfico por molecular en familiares con alta sospecha.
Diagnóstico prenatal ...
Por todo esto tiene gran importancia de la colaboración estrecha entre urólogos, nefrólogos y genetistas.
Conclusiones
El RVU primario se puede presentar de forma aislada o asociado a síndromes con alteraciones extrarrenales (estos últimos con herencia mendeliana reconocida) y presenta agrupación familiar.
En el caso del RVU aislado, el tipaje de los genes es complicado y es tarea de investigación en el momento actual habiendo una serie de propuestas que en la mayoría de los casos apuntan al carácter poligénico de dicho fenómeno.
Es de gran interés el estudio de los factores que determinan la posición de la YU en el desarrollo embriológico.
Los pacientes afectos de RVU han de ser evaluados de forma global por la posibilidad de presentar patología extrarrenal e indagar en los antecedentes personales y familiares. Hoy día no hay criterios objetivos para la selección de pacientes subsidiarios de estudio.
El estudio genético se debe realizar en caso de RVU sindrómico con mutación conocida, expresividad fija, penetrancia completa así como en aquellos casos de afectación grave que comprometa la vida.
Los avances en el campo de la embriología y la regulación de la misma sugieren que la NR congénita no es consecuencia de una mala evolución de los pacientes afectos de RVU sino que se trata de un proceso común por fallo en el desarrollo embrionario.
Es necesaria una estrecha relación entre profesionales de la salud de modo que las novedades en el campo de investigación (como puede ser el análisis proteómico de la orina) sean de aplicación en el terreno práctico una vez avalada su utilidad y con las precauciones oportunas.
Bibliografía y lecturas recomendadas (*lectura de interés y ** lectura fundamental)
*1. MAK, R.H.; KUO, H.J.: Primary ureteral reflux: emerging insights from molecular and genetic studies. Curr. Opin. Pediatr., 15: 181, 2003. [ Links ]
2. BURGER, R.H.; SMITH, C.: Hereditary and familial vesicoureteral reflux. J. Urol., 106: 845, 1971. [ Links ]
**3. CHAPMAN, C.J.; BAILEY, R.R.; JANUS, E.D. y cols.: Vesicoureteric reflux: Segregation analysis. Am. J. Med. Genet., 20: 577, 1985. [ Links ]
4. http://spitfire.emmes.com/study/ped/resources/annlrept2004.pdf, NAPRTCS Annual Report, 2004. [ Links ]
5. http://secure.cihi.ca/cihiweb/dispPage.jsp?cw_page=PG_31_E&cw_topic=31&cw_rel=AR_5_E. CORR Annual Report. Dialysis and Renal Transplantation, 2001. [ Links ]
6. WINGEN, A.M.; FABIAN-BACH, C.; SCHAEFER, F. y cols.: Randomised multicentre study of a low-protein diet on the progression of chronic renal failure in children. European Study Group of Nutritional Treatment of Chronic Renal Failure in Childhood. Lancet, 349: 1117, 1997. [ Links ]
7. WUHL, E.; MEHLS, O.; SCHAEFER, F.; ESCAPE Trial Group: Antihypertensive and antiproteinuric efficacy of ramipril in children with chronic renal failure. Kidney Int., 66: 768, 2004. [ Links ]
8. KING, L.R.: Vesicoureteral reflux: A radiographic sign common to multiple diseases. JAMA, 220: 854, 1972. [ Links ]
9. KAEFER, M.; CURRAN, M.; TREVES, S.T. y cols.: Sibling vesicoureteral reflux in multiple gestation births. Pediatrics, 105: 800, 2000. [ Links ]
10. TOBENKIN, M.I.: Hereditary vesicoureteral reflux. South Med. J., 57: 139, 1964. [ Links ]
11. DEVRIENDT, K.; GROENEN, P.; VAN ESCH, H. y cols.: Vesicoureteral reflux: A genetic condition?. Eur. J. Pediatr., 157: 265, 1998. [ Links ]
12. PASCH, A.; HOEFELE, J.; GRIMMINGER, H. y cols.: Multiple urinary tract malformations with likely recessive inheritance in a large Somalian kindred. Nephrol. Dial Transplant., 19: 3172, 2004. [ Links ]
13. MIDDLETON, G.W.; HOWARDS, S.S.; GILLENWATER, J.Y.: Sex-linked familial reflux. J. Urol., 114: 36, 1975. [ Links ]
*14. BURGER, R.H.: Familial and hereditary vesicouretral reflux. Jama, 216: 680, 1971. [ Links ]
15. DE VARGAS, A.; EVANS, K.; RANSLEY, P. y cols.: A family study of vesicoureteric reflux. J. Med. Genet., 15: 85, 1978. [ Links ]
*16. HEALE, W.F.: Hereditary vesicoureteric reflux: Phenotypic variation and family screening. Pediatr. Nephrol.,11: 504, 1997. [ Links ]
*17. JERKINS, G.R.; NOE, H.N.: Familial vesicoureteral reflux: A prospective study. J. Urol., 128: 774, 1982. [ Links ]
18. NOE, H.N.: The long-term results of prospective sibling reflux screening. J. Urol., 148: 1739, 1992. [ Links ]
19. BAILEY, R.R.; JANUS, E.; McLOUGHLIN, K. y cols.: Familial and genetic data in reflux nephropathy. Contrib. Nephrol., 39: 40, 1984. [ Links ]
20. BAILEY, R.R.: Long-term follow-up of infants with gross vesico-ureteric reflux. Contrib. Nephrol., 39: 146, 1984. [ Links ]
21. CAIONE, P.; VILLA, M.; CAPOZZA, N. y cols.: Predictive risk factors for chronic renal failure in primary high-grade vesicoureteric reflux. BJU Int., 93: 1309, 2004. [ Links ]
22. SMELLIE, J.; EDWARDS, D.; HUNTER, N. y cols.: Vesico-uretericreflux and renal scarring. Kidney Int. Suppl., 4: 65, 1975. [ Links ]
23. BATOURINA, E.; CHOI, C.; PARAGAS, N. y cols.: Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nat. Genet., 32: 109, 2002. [ Links ]
*24. SCHWARZ, R.D.; STEPHENS, F.D.; CUSSEN, L.J.: The pathogenesis of renal dysplasia. II. The significance of lateral and medial ectopy of the ureteric orifice. Invest. Urol., 19: 97, 1981. [ Links ]
25. ICHIKAWA, I.; KUWAYAMA, F.; POPE, J.C. 4th. y cols.: Paradigm shift from classic anatomic theories to contemporary cell biological views of CAKUT. Kidney Int., 61: 889, 2002. [ Links ]
**26. MACKIE, G.G.; STEPHENS, F.D.: Duplex kidneys: A correlation of renal dysplasia with position of the ureteral orifice. J. Urol., 114: 274, 1975. [ Links ]
**27. MURER, L.: Embryology and genetics of primary vesico-ureteric reflux and associated renal dysplasia. Pediatr. Nephrol., 22: 788, 2007. [ Links ]
**28. MURAWSKI, I.J.; GUPTA, I.R.: Vesicoureteric reflux and renal malformations: A developmental problem. Clin. Genet., 69: 105, 2006. [ Links ]
**29. WEBER, S.; MORINIERE, V.; KNUPPEL, T. y cols.: Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: Results of the ESCAPE study. J. Am. Soc. Nephrol., 17: 2864, 2006. [ Links ]
30. SENGAR, D.P.; McLEISH, W.A.; RASHID, A. y cols.: Histocompatibility antigens in urinary tract infection and vesicoureteral reflux: A preliminary communication. Clin. Nephrol., 10: 166, 1978. [ Links ]
31. SENGAR, D.P.; RASHID, A.; WOLFISH, N.M.: Familial urinary tract anomalies: Association with the major histocompatibility complex in man. J. Urol., 121: 194, 1979. [ Links ]
32. MACKINTOSH, P.; ALMARHOOS, G.; HEATH, D.A.: HLA linkage with familial vesicoureteral reflux and familial pelviureteric junction obstruction. Tissue Antigens, 34: 185, 1989. [ Links ]
33. TORRES, V.E.; MOORE, S.B.; KURTZ, S.B. y cols.: In search of marker for genetic susceptibility to reflux nephropathy. Clin. Nephrol., 14: 217, 1980. [ Links ]
34. JENKINS, D.; BITNER-GLINDZICZ, M.; MALCOLM, S. y cols.: De novo uroplakin IIIa heterozygous mutations cause renal adysplasia leading to severe kidney failure. J. Am. Soc. Nephrol., 16: 2141, 2005. [ Links ]
35. LU, W.; PETERS, R.; FERGUSON, H. y cols.: Disruption of ROBO2 is associated with vesicoureteral reflux. J. Am. Soc. Nephrol., 15: 32, 2004. [ Links ]
36. OSHIMA, K.; MIYAZAKI, Y.; BROCK, J.W. III y cols.: Angiotensin type II receptor expression and ureteral budding. J. Urol., 166: 1848 2001. [ Links ]
**37. FEATHER, S.A.; MALCOLM, S.; WOOLF, A.S. y cols.: Primary, nonsyndromic vesicoureteric reflux and its nephropathy is genetically heterogeneous, with a locus on chromosome 1. Am. J. Hum. Genet., 66: 1420, 2000. [ Links ]
38. FAVOR, J.; SANDULACHE, R.; NEUHAUSER-KLAUS, A. y cols.: The mouse Pax2(1Neu) mutation is identical to a human PAX2 mutation in a family with renal-coloboma syndrome and results in developmental defects of the brain, ear, eye, and kidney. Proc. Natl. Acad. Sci. USA, 93: 13870, 1996. [ Links ]
39. DENG, F.M.; DING, M.; LEVKER, R.M. y cols.: Urothelial function reconsidered: A role in urinary protein secretion. Proc. Natl. Acad. Sci. USA, 98: 154, 2001. [ Links ]
40. KERR, D.E.; LIANG, F.X.; BONDIOLI, K.R. y cols.: The bladder as a bioreactor: Urothelium production and secretion of growth hormone into urine. Nat. Biotechnol., 16: 75, 1998. [ Links ]
**41. MAK, R.H.; LITTLE, B.; SKOOG, S. y cols.: Identification of specific biomarkers in children with primary vesico-ureteric reflux by a proteomics approach. Am. Soc. Nephrol., 11: 1983, 2000. [ Links ]
*42. WOOLF, A.S.: Renal Hypoplasia and Dysplasia: Starting to Put the Puzzle Together. J. Am. Soc. Nephrol., 17: 2647, 2006. [ Links ]
*43. CRAIG, J.C.; IRWIG, L.M.; KNIGHT, J.F. y cols.: Does treatment of vesicoureteric reflux in childhood prevent end-stage renal disease attributable to reflux nephropathy?. Pediatrics, 105: 1236, 2000. [ Links ]
*44. WHEELER, D.; VIMALACHANDRA, D.: Antibiotics and surgery for vesicoureteric reflux: A meta-analysis of randomised controlled trials. Arch. Dis. Child., 88: 688, 2003. [ Links ]
*45. GARIN, E.H.: Clinical significance of primary vesicoureteral reflux and urinary antibiotic prophylaxis after acute pyelonephritis: A multicenter, randomized, controlled study. Pediatrics, 117: 626, 2006. [ Links ]
46. MOORTHY, I.: The presence of vesicoureteric reflux does not identify a population at risk for renal scarring following a first urinary tract infection. Arch. Dis. Child, 90: 733, 2005. [ Links ]
47. GRMEK, M.; FETTICH, J.: The importance of follow-up of children with vesicoureteral reflux grade 1. Acta Paediatr., 92: 435, 2003. [ Links ]
48. ARESES, R. y cols.: Reflujo vesicoureteral primario severo en el primer año de vida. Revisión de nuestra casuística. An. Pediatr. Barc., 61: 87, 2004. [ Links ]
49. NOE, H.N.: The long-term results of prospective sibling reflux screening. J. Urol., 148: 1739, 1992. [ Links ]
50. GILTAY, J.C.; VAN DE MEERAKKER, J.; VAN AMSTEL, H.K. y cols.: No pathogenic mutations in the uroplakin III gene of 25 patients with primary vesicoureteral reflux. J. Urol., 171: 931, 2004. [ Links ]
51. KELLY, H.; ENNIS, S.; YONEDA, A. y cols.: Uroplakin III is not a major candidate gene for primary vesicoureteral reflux. Eur. J. Hum. Genet., 13: 500, 2005. [ Links ]
52. OHTOMO, Y.; NAGAOKA, R.; KANEKO, K. y cols.: Angiotensin converting enzyme gene polymorphism in primary vesicoureteral reflux. Pediatr. Nephrol., 16: 648, 2001. [ Links ]
53. RIGOLI, L.; CHIMENZ, R.; DI BELLA, C. y cols.: Angiotensin converting enzyme and angiotensin type 2 receptor gene genotype distributions in Italian children with congenital uropathies. Pediatr. Res., 56: 988, 2004. [ Links ]
*54. YONEDA, A.; OUE, T.; PURI, P.: Angiotensin-converting enzyme genotype distribution in familial vesicoureteral reflux. Pediatr. Surg. Int., 17: 308, 2001. [ Links ]
55. PARK, H.W.; KOO, J.W.; KIM, J.S. y cols.: Association of angiotensin I converting enzyme gene polymorphism with reflux nephropathy in children. Nephron., 86: 52, 2000. [ Links ]
56. HOHENFELLNER, K.; HUNLEY, T.E.; YERKES, E. y cols.: Angiotensin II, type 2 receptor in the development of vesico-ureteric reflux. BJU Int., 83: 318, 1999. [ Links ]
*57. YONEDA, A.; CASCIO, S.; GREEN, A. y cols.: Angiotensin II type receptor gene is not responsible for familial vesicoureteral reflux. J. Urol., 168: 1138, 2002. [ Links ]
58. JIANG, S.; GITLIN, J.; DENG, F.M. y cols.: Lack of major involvement of human uroplakin genes in vesicoureteral reflux: Implications for disease heterogeneity. Kidney Int., 66, 2004. [ Links ]
59. NISHIMURA, H.; YERKES, E.; HOHENFELLNER, K. y cols.: Role of the angiotensin type 2 receptor gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Mol. Cell.; 3: 1, 1999. [ Links ]
60. HIRAOKA, M.; TANIGUCHI, T.; NAKAI, H. y cols.: No evidence for AT2R gene derangement in human urinary tract anomalies. Kidney. Int., 59: 1244, 2001. [ Links ]
61. NAKANO, T.; NIIMURA, F.; HOHENFELLNER, K. y cols.: Screening for mutations in BMP4 and FOXC1 genes in congenital anomalies of the kidney and urinary tract in humans. Tokai. J. Exp. Clin. Med., 28: 121, 2003. [ Links ]
**62. SANNA-CHERCHI, S.; REESE, A.; HENSLE, T. y cols.: Familial vesicoureteral reflux: Testing replication of linkage in seven new multigenerational kindreds. J. Am. Soc. Nephrol., 2005. [ Links ]
63. SHEFELBINE, S.E.; KHORANA, S.; SCHULTZ, P.N. y cols.: Mutational analysis of the GDNF/RET-GDNFR alpha signalling complex in a kindred with vesicoureteral reflux. Hum. Genet., 102: 474, 1998. [ Links ]
64. ZAGRADISNIK, B.; BRACIC, K.; VARDA, N.M. y cols.: G-protein beta3 subunit gene C825T polymorphism in patients with vesico-ureteric reflux. Ann. Genet., 47: 209, 2004. [ Links ]
65. CHOI, K.L.; McNOE, L.A.; FRENCH, M.C. y cols.: Absence of PAX2 gene mutations in patients with primary familial vesicoureteric reflux. J. Med. Genet., 35: 33, 1998. [ Links ]
66. LIPSON, A.H.; YUILLE, D.; ANGEL, M. y cols.: Velocardiofacial (Shprintzen) syndrome: An important syndrome for the dysmorphologist to recognise. J. Med. Genet., 28: 596, 1991. [ Links ]
67. ANDERSON, N.G.; ABBOTT, G.D.; MOGRIDGE, N. y cols.: Vesicoureteric reflux in the newborn: Relationship to fetal renal pelvic diameter. Pediatr. Nephrol., 11: 610, 1997. [ Links ]
68. OGATA, T.; MUROYA, K.; SASAGAWA, I. y cols.: Genetic evidence for a novel gene(s) involved in urogenital development on 10q26. Kidney. Int., 58: 2281, 2000. [ Links ]
69. GORINATI, M.; ZAMBONI, G.; PADOIN, N. y cols.: Terminal deletion of the long arm of chromosome 10: Case report and review of the literature. Am. J. Med. Genet., 33: 502, 1989. [ Links ]
70. LEE-CHEN, G.J.; LIU, K.P.; LAI, Y.C. y cols.: Significance of the tissue kallikrein promoter and transforming growth factor-beta1 polymorphisms with renal progression in children with vesicoureteral reflux. Kidney. Int., 65: 1467, 2004. [ Links ]
71. ANDREU, N.; ESCARCELLER, M.; FEATHER, S. y cols.: PALML, a novel paralemmin-related gene mapping on human chromosome 1p21. Gene., 278: 33, 2001. [ Links ]
72. CHOI, K.L.; McNOE, L.A.; FRENCH, M.C. y cols.: Absence of PAX2 gene mutations in patients with primary familial vesicoureteric reflux. J. Med. Genet., 35: 338, 1998. [ Links ]
73. JENKINS, D.; BITNER-GLINDZICZ, M.; MALCOLM, S. y cols.: De novo uroplakin IIIa heterozygous mutations cause renal adysplasia leading to severe kidney failure. J. Am. Soc. Nephrol., 16: 2141, 2005. [ Links ]
74. MOERMAN, P.; FRYNS, J.P.; SASTROWIJOTO, S.H. y cols.: Hereditary renal adysplasia: New observations and hypotheses. Pediatr. Pathol., 14: 405, 1994. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Antonio Morales Martínez
Avda. San Isidro, 12 6º B - I
29018. Málaga. (España).
morimorales2003@hotmail.com

