










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActas Urológicas Españolas
versión impresa ISSN 0210-4806
Actas Urol Esp vol.32 no.8 sep. 2008
El espectro del carcinoma renal papilar
Papillary renal cell carcinoma spectrum
Ugalde A., López J.I.*
Servicio de Anatomía Patológica. Hospital de Basurto. Bilbao (Vizcaya)
*Departamento de Anatomía Patológica. Hospital de Cruces. Universidad del País Vasco (EHU/UPV). Baracaldo (Vizcaya).
Dirección para correspondencia
RESUMEN
Objetivos: Los últimos años están asistiendo a la gran expansión conceptual del cáncer renal. La conjunción entre los hallazgos histológicos y los datos moleculares ha sido un punto clave en este fenómeno, y resultado de ello ha sido la clasificación de los tumores renales del adulto de la OMS de 2004. En ella se han unido viejos y nuevos tumores renales. Lejos de estar cerrada, esta clasificación se actualiza gracias a nuevos puntos de vista surgidos a la luz de los nuevos hallazgos que se obtienen fundamentalmente en el campo de la genética.
Material y Métodos: El carcinoma papilar renal en el sentido más amplio del término es el objeto de revisión en este trabajo. Este fenotipo histológico en el cáncer renal es un buen ejemplo de evolución incesante de la anatomía patológica como disciplina clínica, con un crecimiento importante en los últimos tiempos.
Resultados: El conjunto de neoplasias renales que muestran una histología papilar o túbulo-papilar, o que tienen su sustrato genético, algunas de las cuales son entidades muy recientes y están aún hoy en día en discusión, es amplio y variado: carcinoma papilar esporádico convencional, carcinomas papilares ligados a síndromes genéticos (carcinoma papilar hereditario, carcinoma papilar asociado a leiomiomatosis hereditaria, carcinoma papilar asociado a carcinoma papilar hereditario de tiroides, síndrome de Birt-Hogg-Dubé) o a alteraciones genéticas específicas (carcinomas papilares con translocación Xp11.2), carcinomas papilares con morfología peculiar (carcinoma micropapilar, carcinoma papilar invertido, carcinoma papilar con células fusiformes y túbulos angulados) y carcinomas renales de nuevo cuño englobados dentro del grupo del carcinoma papilar (carcinoma tubulo-quístico y carcinoma mucinoso, tubular y fusocelular).
Conclusión: Además de las variedades clásicamente reconocidas, nuevas entidades, algunas no bien definidas aún, están engrosando de forma progresiva el grupo de carcinomas renales con morfología papilar. Lejos de agotarse, este crecimiento continuará en los próximos años a la luz de los nuevos hallazgos, y en éstos los patólogos tendrán mucho que decir.
Palabras clave: Tumor renal. Clasificación de la OMS. Carcinoma papilar. Alteración genética. Histología. Diagnóstico diferencial.
ABSTRACT
Objectives: Significant conceptual expansion of renal cancer continues to increase. The key point to this phenomenon is based on the combination of morphology and molecular data. The result is the new 2004 WHO classification of renal tumors in adults. The apparently never ending advance in molecular genetics is constantly pushing to update recently proposed listings.
Material and Methods: Papillary renal cell carcinoma, considered the term in the broader sense, is the subject of this study. This histological phenotype in renal cancer, with an accelerated growth in the last times, is a good example of the never ending evolution of pathology as a clinical discipline.
Results: The genetic background and the phenotype of all renal neoplasms with papillary or tubulo-papillary phenotype, or with its genetic background, some of them being very recently described entities even now under discussion is wide and heterogenous: conventional sporadic papillary carcinoma, papillary carcinomas linked to genetic syndromes (hereditary papillary carcinoma, papillary carcinoma associated to hereditary leiomyomatosis, papillary carcinoma associated to hereditary papillary thyroid carcinoma, Birt-Hogg-Dubé syndrome) or to specific genetic disorders (Xp11.2 associated papillary carcinoma), papillary carcinoma with distinct morphology (micropapillary carcinoma, inverted papillary carcinoma, papillary carcinoma with spindle cells and angulated tubules) and new renal carcinomas included within the group of papillary carcinoma (tubulo-cystic carcinoma and tubular, mucinous and spindle cell carcinoma).
Conclusion: Aside from the classically known histological variants, several new entities, some of them still badly delineated, are progressively enlarging the group of renal carcinomas with papillary phenotype. This growth will continue in the next times on the light of the new findings and pathologists will be main actors in this fact.
Key words: Renal tumor. WHO classification. Papillary carcinoma. Genetic disorder. Histology. Differential diagnosis.
La clasificación de la OMS publicada en 2004 reconoce 49 tipos diferentes de neoplasia renal1. Aproximadamente entre el 10 y el 15% de los carcinomas de células renales son carcinomas papilares (CCRP)2,3. La primera descripción detallada de esta variedad histológica de cáncer renal apareció en 19764. Años después, Thoenes et al.5 los denominó en su clasificación carcinomas renales cromófilos debido a la basofilia citoplásmica que exhibían con las técnicas convencionales de tinción. Sin embargo, esta terminología fue abandonada con rapidez debido a que el término resultaba equívoco, puesto que sólo una parte de los CCRP tienen el citoplasma basófilo. En 1993, la clasificación de Mainz6 comenzó a integrar los primeros hallazgos citogenéticos y supuso un nuevo paso hacia adelante en el conocimiento de estos tumores. Más tarde, Delahunt y Eble7 distinguieron dos variantes histológicas del CCRP, que ellos denominaron Tipos 1 y 2. Esta subclasificación se vio refrendada poco tiempo después en base a estudios moleculares8.
La inmensa mayor parte de los CCRP son de presentación esporádica. Sin embargo, se han descrito casos hereditarios9, y casos asociados a otros tumores como leiomiomatosis hereditaria10, carcinoma papilar de tiroides hereditario11, y síndrome de Birt-Hogg-Dubé12. Además, se han descrito casos de carcinoma renal de morfología papilar en la edad pediátrica asociados a la translocación Xp11.213. Estudios recientes indican que tanto el carcinoma renal tubular, mucinoso, y fusocelular14-20 como el carcinoma renal túbulo-quístico21-24 comparten las mismas alteraciones cromosómicas que el CCRP, lo cual los convierte, según varios autores18,20 en nuevas variantes de CCRP. Existe, sin embargo, cierta controversia al respecto19. Muy recientemente se han presentado nuevas variedades de CCRP, por ejemplo, el carcinoma papilar con células fusiformes de bajo grado y túbulos angulados25 que remeda el carcinoma tubular, mucinoso y fusocelular, el carcinoma papilar renal invertido26 y el carcinoma renal micropapilar26.
Este trabajo pasa revista a los aspectos clínicos del CCRP, así como el creciente espectro histológico y molecular de esta variedad de cáncer renal, incluyendo en el mismo a todas las variantes últimamente propuestas.
Carcinoma renal papilar esporádico
La presentación esporádica es, con mucho, la forma más común de CCRP. Muestran cierta tendencia a la multifocalidad, lo cual no contraindica la cirugía conservadora27. En ocasiones se asocian a adenomas, e incluso se han descrito casos de CCRP múltiples con nódulos de histología mixta, a caballo entre adenoma y carcinoma, lo cual a algunos autores28 les sugiere una transición entre ambos.
Los CCRP son neoplasias generalmente bien circunscritas y con frecuencia excéntricas en el riñón. A pesar de ello, la mayor parte están confinadas en el órgano y muestran cápsula2. Al corte muestran un aspecto muy heterogéneo, dependiendo del grado de necrosis y de la extensión de la hemorragia, ya que ambos fenómenos son muy característicos de este tumor. La necrosis puede ser lo suficientemente extensa como para ser interpretado en los estudios radiológicos como quiste en vez de como un tumor. El patólogo, en estos casos extremos, debe buscar, habitualmente en la periferia, las áreas mejor conservadas para poder emitir un diagnóstico. A pesar de su notoriedad, se ha demostrado recientemente que la extensión de la necrosis no influye en el pronóstico29.
Desde que Delahunt y Eble publicaron su artículo en 1997 (7) se distinguen 2 tipos histológicos bien definidos de CCRP. El tipo 1 (Fig. 1) es el más frecuente y el que tiene mejor pronóstico. Está constituido por células pequeñas, con escaso citoplasma basófilo, núcleo redondeado, y nucleolo poco aparente que se disponen en hilera simple en los ejes hialino-vasculares de las papilas, en los cuales aparecen con frecuencia acúmulos de histiocitos espumosos y, ocasionalmente, cuerpos de psammoma. Se trata de tumores de bajo grado. El tipo 2 (Fig. 2), menos frecuente, se caracteriza por tener papilas irregulares y células de gran tamaño con citoplasma amplio y eosinófilo, y núcleo voluminoso con nucleolo notorio. Los acúmulos histiocitarios y los cuerpos de psammoma aparecen con menos frecuencia. El grado nuclear es mayor en el tipo 2 que en el tipo 130. Recientemente se ha propuesto el grado nucleolar31 como el más adecuado para estos tumores en vez del grado de Fuhrman convencional. Asimismo, el estadio TNM32 es más avanzado en el CCRP de tipo 2.


Al igual que el resto de los carcinomas renales, el CCRP puede presentar transformación sarcomatoide33. En ocasiones es de morfología sólida34, o mixta, tubular y papilar y, más raramente, de predominio tubular y en estos casos pueden presentarse dificultades diagnósticas. En muy raras ocasiones35, sus células presentan cambios oncocíticos.
El perfil inmunohistoquímico del CCRP incluye positividades para CD10, marcador del carcinoma renal, queratina 7, y p504S. La queratina 20 y las queratinas de alto peso molecular son negativas. El tipo 1 es positivo, además, con MUC136 y el tipo 2 con e-cadherina37. Desde el punto de vista molecular, el CCRP se caracteriza por presentar trisomías o tetrasomías de los cromosomas 7 y 17 y pérdida del cromosoma Y6,38. La trisomía 7 es característica pero no específica, pudiendo encontrarse también en tumores de vejiga, próstata y de cabeza y cuello, en la hiperplasia benigna de próstata y en otras enfermedades benignas. Sin embargo, es una alteración muy constante y frecuente en las fases tempranas de la enfermedad, lo que sugiere que los genes situados en él juegan un papel crítico en la patogenia de estos tumores39. La trisomía 17 es mucho más específica y valorable desde el punto de vista diagnóstico, pudiendo encontrarse trisomías completas, isocromosomas 17q, o duplicaciones de 17q21. El análisis molecular del CCRP también tiene implicaciones pronósticas. Así, los casos con pérdida de Xp (8) o de 9p40 tienen mal pronóstico con supervivencias acortadas. Por último, Gunawan et al.40 demuestran que el tipo 2 surge por evolución del tipo 1, conectando a nivel molecular ambas variedades de CCRP.
Carcinoma renal papilar hereditario
El CCRP hereditario es una enfermedad rara, autosómica dominante, que se caracteriza por presentar tumores renales bilaterales múltiples de comienzo tardío, aunque existen casos aislados de aparición temprana41. El tumor presenta una evolución indolente, hasta el punto de que muchos pacientes son asíntomaticos en el momento del diagnóstico y éste se realiza al descubrirse los tumores en exámenes radiológicos realizados de rutina, o por otras causas.
Histológicamente la enfermedad se caracteriza por presentar múltiples tumores renales de morfología papilar o túbulo-papilar, observándose toda la gama imaginable, desde tumores microscópicos diseminados a lo largo del parénquima renal hasta carcinomas papilares de tipo 1, pasando por adenomas papilares y tumores híbridos42. Las células tumorales son típicamente de bajo grado. Curiosamente, hasta el 70% de los CCRP hereditarios contienen células claras (42), en especial aquellas áreas que se acompañan de necrosis o hemorragia.
El CCRP hereditario está causado por mutaciones en el proto-oncogén c-MET, que se localiza en 7q31-3443,44. La sobre-expresión de c-MET, al igual que la de proto-oncogenes análogos como c-kit y RET, está asociado a múltiples neoplasias45. Las alteraciones responsables del CCRP hereditario suele ser bien mutaciones en línea germinal a nivel de los exones 16 a 19, o bien trisomías 7. Un pequeño grupo de CCRP esporádicos también muestra mutaciones de c-MET idénticas a las de las formas hereditarias42,43.
Carcinoma renal papilar asociado a leiomiomatosis hereditaria
El síndrome de leiomiomatosis hereditaria y carcinoma renal asocia leiomiomas cutáneos y uterinos junto a carcinoma renal papilar de tipo 2, y fue descrito en tres familias en 200110,46. Aproximadamente el 30% de los pacientes con este síndrome desarrollan un único carcinoma renal de alto grado de malignidad histológicamente superponible al tipo 2 de carcinoma papilar renal47, aunque se han descrito casos morfológicamente superponibles al carcinoma de ductos colectores y oncocitomas. Se trata de tumores de alta agresividad biológica, con elevado índice de metástasis en el momento del diagnóstico. El síndrome se transmite de manera autosómica dominante y la alteración se localiza en 1q42.3-4347.
Histológicamente muestra una arquitectura papilar o túbulo-papilar con pleomorfismo nuclear y nucleolos eosinófilos con halo perinucleolar característico (Fig. 3). Los citoplasmas son amplios y eosinófilos y cuando forman túbulos, las luces contienen un material basófilo.

Carcinoma renal papilar asociado a carcinoma papilar de tiroides hereditario
Se ha descrito recientemente una familia con carcinoma papilar de tiroides hereditario en la cual dos miembros desarrollaron un carcinoma papilar renal con mutación en c-MET similar a la del CCRP hereditario descrito previamente11. Sin embargo, la alteración genética que liga los carcinomas papilares de ambos órganos parece estar localizada en 1q2111, región cromosómica en la que se localizan varios oncogenes, entre ellos N-RAS, NTRK-1 (codifica una tirosin-kinasa implicada en el carcinoma papilar de tiroides) y PRCC (oncogén implicado en el carcinoma renal)48.
Carcinoma renal asociado al síndrome de birt-hogg-dube
El síndrome de Birt-Hogg-Dubé es una enfermedad autosómica dominante que consiste en una triada de lesiones cutáneas compuesta por fibrofoliculomas, tricodiscomas, y acrocordones49, al que se añaden con cierta frecuencia neumotórax espontáneos, neoplasias colorectales, y diversos tipos de carcinoma renal entre los que predomina el carcinoma de células cromófobas50. Más raramente se han descrito carcinomas renales papilares que corresponden histológicamente al tipo 251. La alteración genética de este síndrome se localiza en el cromosoma 1712.
Carcinoma renal con translocación Xp11
Bajo este nombre se agrupan un grupo de carcinomas renales que se presentan en la edad pediátrica y que tienen en común una translocación en la que el punto de rotura está en Xp11.2. Algunos de estos pacientes se caracterizan por haber recibido varios años antes tratamientos de quimioterapia citotóxica52. La fusión ASPL-TFE3 es la que acompaña al tumor renal de morfología más típicamente papilar. El tumor resultante es de curso indolente a pesar de presentarse en estadios avanzados y en edades tempranas. Recientemente se han descrito carcinomas renales con translocación Xp11.2 en la edad adulta53.
Desde el punto de vista histológico, los carcinomas renales con translocación Xp11.2 están compuestos por papilas tapizadas por células claras, así como por nidos y alvéolos acompañados por cuerpos de psammoma. A veces presentan citoplasmas levemente eosinófilos. Estos tumores son característicamente positivos con marcadores inmunohistoquímicos de melanoma (Melan-A, HMB45) y no expresan, o lo hacen muy débilmente, citoqueratinas y vimentina52,54. Asimismo, son positivos con CD10 y con el marcador de carcinoma renal. De manera específica estos tumores muestran tinción nuclear con TFE354.
Como consecuencia de la rotura en Xp11.2 se producen fusiones cromosómicas entre el factor de transcripción TFE3, codificado en la región de la rotura, y otros genes54,55. Entre ellas, la ASPL-TFE3 es idéntica a la que se produce en el sarcoma alveolar de partes blandas55.
Carcinoma papilar renal invertido
El CCRP invertido se ha definido muy recientemente como aquel carcinoma papilar en el que los núcleos de las células se localizan en el lado opuesto al convencional, es decir, en la superficie luminar de la papila, con citoplasmas eosinófilos intensos en situación basal. El patrón inmunohistoquímico es peculiar, con positividad para MUC1, e-cadherina, y CD117 y negatividad para Ki67 y CK20. La importancia de reconocer esta variedad estriba en su supuesto mejor pronóstico26.
Carcinoma renal tubular, mucinoso y fusocelular
La primera referencia a este tumor bajo esta denominación corrió a cargo de Rakozy et al. en 200214. Desde esa fecha se han publicado varias series15-20 y todas ellas coinciden en que se trata de un tumor renal de bajo grado de malignidad.
El carcinoma renal tubular, mucinoso y fusocelular muestra una histología muy específica14-20 (Fig. 4). El patrón predominante consiste en agrupaciones de células fusiformes que engloban cordones, túbulos, y papilas de células cuboideas, todas ellas con escasa atipia, monótonas y eosinófilas. El estroma es de aspecto mixoide y muestra vacuolas de aspecto mucinoso que se tiñe con el azul Alcian. De forma característica, se observan grupos de histiocitos espumosos en el estroma.

Existe una cierta controversia respecto de la naturaleza de este tumor renal. Por un lado, Paner et al.18 llaman la atención sobre las similitudes de este tumor con el CCRP, opinión que ha sido reforzada más recientemente20. Sin embargo, Cossu-Rocca et al.19 encuentran diferencias sustanciales a nivel genético entre ambos tumores, por lo cual consideran que el carcinoma renal mucinoso, tubular y fusocelular es una entidad distinta del carcinoma papilar convencional.
Carcinoma renal micropapilar
EL CCRP variante micropapilar está compuesto por papilas pequeñas, a veces sin eje conectivo vascular, frágiles, que aparecen muchas veces flotando en las luces. El tumor remeda histológicamente al carcinoma seroso de la superficie ovárica. El inmunofenotipo es similar al del CCRP convencional. Al igual que ocurre con los carcinomas micropapilares de otros órganos, el CCRP micropapilar tiene un comportamiento agresivo26.
Carcinoma renal túbulo-quístico
MacLennan et al.21 describieron en 1997 una nueva variedad de carcinoma renal de bajo grado, que consideraron la variante de bajo grado del carcinoma de los túbulos colectores de Bellini. Se trata de un tumor muy característico desde el punto de vista macroscópico e histológico. Al corte el tumor presenta un aspecto esponjoso, con abundantes microquistes (Fig. 5). Histológicamente el tumor está compuesto por ductos dilatados y tapizados por células de aspecto apocrino con núcleos grandes con nucleolo prominente (Fig. 6). No se observan mitosis.
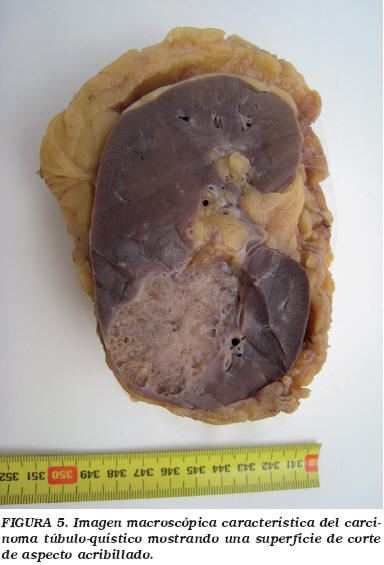

Se trata de un tumor de buen pronóstico. A pesar de haber sido considerado por algunos autores como una nueva entidad22, algunos estudios recientes demuestran un patrón inmunohistoquímico y alteraciones genéticas superponibles al CCRP23,24.
Carcinoma renal con células fusiformes de bajo grado y túbulos angulados
Muy recientemente se han descrito 5 casos de carcinoma papilar de patrón sólido conteniendo escasas papilas y abundantes áreas compactas de células fusiformes englobando túbulos de contornos angulados25. Todos los casos mostraron el típico patrón molecular e inmunohistoquímico del CCRP, con trisomias 7 y 17 y positividad para CK 725.
Conclusiones
El CCRP es una variedad histológica frecuente de cáncer renal cuyo espectro histológico está ampliándose en los últimos tiempos gracias fundamentalmente a la aportación de la biología molecular. Este hecho es una muestra más de la importancia de esta técnica diagnóstica en el conocimiento del cáncer. En el terreno del cáncer renal ha propiciado una evolución trepidante en el conocimiento de esta patología en los últimos años, circunstancia que se ve reflejada en la última clasificación de la OMS de 2004.
Referencias
1. Eble JN, Sauter G, Epstein JI. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Lyon, IARC Press, 2004. [ Links ]
2. Murphy WM, Grignon DJ, Perlman EJ. Tumors of the Kidney, Bladder, and Related Urinary Structures. AFIP Atlas of Tumor Pathology, 4th Series, Fascicle 1. American Registry of Pathology, Washington DC, 2004. [ Links ]
3. López-Beltrán A, Scarpelli M, Montironi R, Kirkali Z. 2004 WHO classification of the renal tumors of the adults. Eur Urol. 2006; 49(5):798-805. [ Links ]
4. Mancilla-Jimenez R, Stanley RJ, Blath RA. Papillary renal cell carcinoma. A clinical, radiologic and pathologic study of 34 cases. Cancer. 1976;38(6):2469-2480. [ Links ]
5. Thoenes W, Störkel S, Rumpelt HJ. Histopathology and classification of renal cell tumors (adenomas, oncocytomas and carcinomas): The basic cytological and histopathological elements and their use in diagnostics. Pathol Res Pract. 1986;181(2):125-143. [ Links ]
6. Kovacs G. Molecular differential pathology of renal cell tumours. Histopathology. 1993;22(1):1-8. [ Links ]
7. Delahunt B, Eble JN. Papillary renal cell carcinoma: A clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997;10(6):537-544. [ Links ]
8. Jiang F, Richter J, Schraml P, Bubendorf L, Gasser T, Sauter G, et al. Chromosomal imbalances in papillary renal cell carcinoma: genetic differences between histological subtypes. Am J Pathol. 1998;153(5):1467-1473. [ Links ]
9. Cohen D, Zhou M. Molecular genetics of familial renal cell carcinoma syndromes. Clin Lab Med. 2005;25(2):259-277. [ Links ]
10. Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA. 2001;98(6):3387-3392. [ Links ]
11. Malchoff CD, Sarfarazi M, Tendler B, Forouhar F, Whalen G, Joshi V, et al. Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome. J Clin Endocrinol Metab. 2000;85(5): 1758-176. [ Links ]
12. Khoo SK, Bradley M, Wong FK, Hedblad MA, NordenskjöldM, Teh BT. Birt-Hogg-Dubé syndrome: mapping of a novel hereditary neoplasia gene to chromosome 17p12-q11.2. Oncogene. 2001;20(37):5239-5242. [ Links ]
13. Argani P, Antonescu CR, Couturier J, Fournet JC, Sciot R, Debiec-Rychter M, et al. PRCC-TFE3 renal cell carcinomas: morphologic, immunohistochemical, ultrastructural and molecular analysis o fan entity associated with the t(X;1)(p11.2; q21). Am J Surg Pathol. 2002,26(12):1553-1566. [ Links ]
14. Rakozy C, Schmahl S, Bogner S, Storkel S. Low-grade tubular-mucinous renal neoplasms: Morphologic, immunohistochemical and genetic features. Mod Pathol. 2002;15(11):1162-1171. [ Links ]
15. Ferlicot S, Allory Y, Comperat E, Mege-Lechevalier F, Dimet S, Sibony M, et al. Mucinous tubular and spindle cell carcinoma: a report of 15 cases and review of the literature. Virchows Arch. 2005;447(6):978-983. [ Links ]
16. Gaafar A, Valentí C, Echevarría C, Laforga JB, López JI. Renal mucinous and tubular spindle cell carcinoma: A clinicopathological study of 4 cases. Ann Saudi Med. 2006;26(6):466-470. [ Links ]
17. Brandal P, Lie AK, Bassarova A, Svindland A, Risberg B, Danielsen H, et al. Genomic aberrations in mucinous tubular and spindle cell renal cell carcinomas. Mod Pathol. 2006;19(2):186-194. [ Links ]
18. Paner GP, Srigley JR, Radhakrishnan A, Cohen C, Skinnider BF, Tickoo SK, et al. Immunohistochemical analysis of mucinous tubular and spindle cell carcinoma and papillary renal cell carcinoma of the kidney: Significant immunophenotypic overlap warrants diagnostic caution. Am J Surg Pathol. 2006;30(1):13-19. [ Links ]
19. Cossu-Rocca P, Eble JN, Delahunt B, Zhang S, Martignoni G, Brunelli M, et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol. 2006;19(4):488-493. [ Links ]
20. Shen SS, Ro JY, Tamboli P, Truong LD, Zhai Q, Jung SJ, et al. Mucinous tubular and spindle cell carcinoma of kidney is probably a variant of papillary renal cell carcinoma with spindle cell features. Ann Diagn Pathol. 2007;11(1):13-21. [ Links ]
21. MacLennan GT, Farrow GM, Bostwick DG. Low-grade collecting duct carcinoma of the kidney: Report of 13 cases of low-grade mucinous tubulo-cystic renal carcinoma of possible collecting duct origin. Urology. 1997; 50(5):679-684. [ Links ]
22. Azoulay S, Vieillefond A, Paraf F, Pasquier D, Cussenot O, Callard P, et al. Tubulocystic carcinoma of the kidney: A new entity among renal tumors. Virchows Arch. 2007;451(5):905-909. [ Links ]
23. Sercia L, Yang XJ, López JI, Hes O, Shen S, Li R, et al. Renal tubulocystic carcinoma is related to papillary renal cell carcinoma: Cytogenetic and histological evidence. Mod Pathol. 2008;21(Suppl):180A. [ Links ]
24. Yang XJ, Zhou M, Hes O, Shen S, Li R, Lopez J, et al. Tubulocystic carcinoma of the kidney: clinicopathologic and molecular characterization. Am J Surg Pathol. 2008;32(2):177-187. [ Links ]
25. Argani P, Netto G, Parwani A. Papillary renal cell carcinoma with low grade spindle cells living angulated tabules: A mimic of mucinous tubular and spindle cell carcinoma. Mod Pathol. 2008;21(Suppl):146A. [ Links ]
26. Park BH, Ro JY, Park WS, Shen S, Cho YM. Papillary renal cell carcinomas with inverted and micropapillary patterns: A clinicopathologic study of 33 cases. Mod Pathol. 2008;21(Suppl):175A. [ Links ]
27. Mejean A, Hopirtean V, Bazin JP, Larousserie F, Benoit H, Chrétien Y, et al. Prognostic factors for the survival of patients with papillary renal cell carcinoma: meaning of histological typing and multifocality. J Urol. 2003;170(3):764-767. [ Links ]
28. Kiyoshima K, Oda Y, Nakamura T, Migita T, Okumura K, Naito S, et al. Multicentric papillary renal cell carcinoma associated with renal adenomatosis. Pathol Int. 2004;54(4):266-272. [ Links ]
29. Lee SE, Byun SS, Oh JK, Lee SC, Chang IH, Choe G, et al. Significance of macroscopic tumor necrosis as a prognostic indicador for renal cell carcinoma. J Urol. 2006;176(4 pt 1):1332-1337. [ Links ]
30. Delahunt B, Eble JN, McCredie MR, Bethwaite PB, Stewart JH, Bilous AM. Morphologic typing of papillary renal cell carcinoma: Comparison of growth kinetics and patient survival in 66 cases. Hum Pathol. 2001;32(6):590-595. [ Links ]
31. Sika-Paotonu D, Bethwaite PB, McCredie MRE, William Jordan T, Delahunt B. Nucleolar grade but not Fuhrman grade is applicable to papillary renal cell carcinoma. Am J Surg Pathol. 2006;30(9):1091-1096. [ Links ]
32. Sobin LH, Wittekind C. TNM classification of Malignant Tumours, 6th edition, New Jersey, John Wiley & Sons, 2002. [ Links ]
33. Cohen RJ, McNeal JE, Susman M, Sellner LN, Iacopetta BJ, Weinstein SL, et al. Sarcomatoid renal cell carcinoma of papillary origin. A case report and cytogenic evaluation. Arch Pathol Lab Med. 2000;124(12):1830-1832. [ Links ]
34. Renshaw AA, Zhang H, Corless CL, Fletcher JA, Pins MR. Solid variants of papillary (chromophil) renal cell carcinoma: clinicopathologic and genetic features. Am J Surg Pathol. 1997;21(10):1203-1209. [ Links ]
35. Lefèvre M, Couturier J, Sibony M, Bazille C, Boyer K, Callard P, et al. Adult papillary renal tumor with oncocytic cells. Clinicopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am J Surg Pathol. 2005;29(12):1576-1581. [ Links ]
36. Leroy X, Zini L, Leteurtre E, Zerimech F, Porchet M, Aubert JP, et al. Morphologic subtyping of papillary renal cell carcinoma: correlation with prognosis and differential expresión of MUC1 between the two subtypes. Mod Pathol. 2002;15(11):1126-1130. [ Links ]
37. Zhou M, Roma A, Magi-Galluzzi C. The usefulness of immunohistochemical markers in the differential diagnosis of renal neoplasms. Clin Lab Med. 2005;25(2):247-257. [ Links ]
38. Kovacs G, Füzesi L, Emanual A, Kung HF. Cytogenetics of papillary renal cell tumors. Genes Chromosomes Cancer. 1991;3(4):249-255. [ Links ]
39. Glukhova L, Lavialle C, Fauvet D, Chudoba I, Danglot G, Angevin E et al. Mapping of the 7q31 subregion common to the small chromosome 7 derivatives from two sporadic papillary renal cell carcinomas: increased copy number and overexpression of the MET proto-oncogene. Oncogene. 2000;19(6):754-761. [ Links ]
40. Gunawan B, von Heydebreck A, Fritsch T, Huber W, Ringert RH, Jakse G, et al. Cytogenetic and morphologic typing of 58 papillary renal cell carcinomas: evidence for a cytogenetic evolution of Type 2 from Type 1 tumors. Cancer Res. 2003;63(19):6200-6205. [ Links ]
41. Schmidt LS, Nickerson ML, Angeloni D, Glenn GM, Walther MM, Albert PS, et al. Early onset hereditary papillary renal carcinoma: germline missense mutations in the tyrosine kinase domain of the met proto-oncogene. J Urol. 2004;172(4 pt 1):1256-1261. [ Links ]
42. Lubensky IA, Schmidt L, Zhuang Z, Weirich G, Pack S, Zambrano M, et al. Hereditary and sporadic papillary renal cell carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999;155(2):517-526. [ Links ]
43. Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16(1):68-73. [ Links ]
44. Bottaro DP, Linehan WM. Multifocal renal cancer: genetic basis and its medical relevance. Clin Cancer Res. 2005;11(20):7206-7208. [ Links ]
45. Di Renzo MF, Narsimhan RP, Olivero M, Bretti S, Giordano S, Medico E, et al. Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene. 1991;6(11):1997-2003. [ Links ]
46. Kiuru M, Launonen V, Hietala M, Aittomaki K, Vierimaa O, Salovaara R, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159(3):825-829. [ Links ]
47. Alam NA, Rowan AJ, Wortham NC, Pollard PJ, Mitchell M, Tyrer JP, et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet. 2003;12(11):1241-1252. [ Links ]
48. Moore FD Jr. Inherited aspects of papillary thyroid carcinoma. J Surg Oncol. 2006;94(88):719-724. [ Links ]
49. Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113(12):1674-1677. [ Links ]
50. Khoo SK, Kahnoski K, Sugimura J. Petillo D, Chen J, Shockley K, et al. Inactivation of BHD in sporadic renal tumors. Cancer Res. 2003;63(15): 4583-4587. [ Links ]
51. Pavlovich CP, Walther MM; Eyler RA, Hewitt SM, Zbar B, Linehan WM, et al. Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol. 2002;26(12):1542-1552. [ Links ]
52. Argani P Lae M, Ballard ET, Amin M, Manivel C, Hutchinson B, et al. Translocation carcinomas of the kidney after chemotherapy in childwood. J Clin Oncol. 2006;24(10):1529-1534. [ Links ]
53. Higa B, Flanigan RC, Picken MM. The incidence of TFE3 translocation renal carcinoma among adult patients. Mod Pathol. 2008;21(Suppl):161A. [ Links ]
54. Argani P, Lae M, Hutchison B, Reuter VE, Collins MH, Perentesis J, et al. Renal carcinomas with the t(6;11)(p21;q12). Clinicopathologic features and demonstration of the specific alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA PCR. Am J Surg Pathol. 2005;29(2): 230-240. [ Links ]
55. Argani P, Hawkins A, Griffin CA, Goldstein JD, Haas M, Beckwith JB, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1; q12) chromosome translocation. Am J Pathol. 2001;158(6): 2089-2096. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. J.I. López
Departamento de Anatomía Patológica. Hospital de Cruces
Plaza de Cruces, s/n - 48903 Baracaldo, Vizcaya
Tel.: 946 006 336
E-mail autor: joseignacio.lopez@ehu.es
Trabajo recibido: abril 2008
Trabajo aceptado: mayo 2008

