










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNefrología (Madrid)
versión On-line ISSN 1989-2284versión impresa ISSN 0211-6995
Nefrología (Madr.) vol.33 no.3 Cantabria 2013
https://dx.doi.org/10.3265/Nefrologia.pre2012.Sep.11558
The first Molecular genetics analysis of individuals suffering from nephropatic cystinosis in the Southwestern Iran
Primer análisis genético molecular en personas que padecen cistinosis nefropática en el sudoeste de Irán
Sepideh Shahkarami1, Hamid Galehdari2, Ali Ahmadzadeh1, Mahnaz Babaahmadi3 and Mohammad Pedram1
1Research Center for Thalassemia and Hemoglobinopathy, Jondishapour University of Medical Sciences. Ahvaz (Iran)
2Department of Genetics, Faculty of Sciences. Shahid Chamran University of Ahvaz. Ahvaz (Iran)
3Toxicology Research Center, Jondishapour University of Medical Sciences. Ahvaz (Iran)
We thank the deputy research center of Ahvaz medical School of Jundishapur University and Research center of thalassemia and hemoglobinopathies of Ahvaz medical School of Jundishapur University for supporting this work.
ABSTRACT
Objective: Nephropatic Cystinosis (NC) is a rare metabolic disorder due to mutation in the CTNS gene in which more than 90 different mutations have already been reported so far. This study was performed to investigate mutations of the CTNS gene and its promoter in a number of Iranian patients with NC.
Methods: Polymerase chain reaction and direct sequencing were performed for molecular characterization of the CTNS gene in 25 patients from 24 unrelated Iranian families with NC.
Results: None of the patients showed the 57kb deletion in heterozygous or homozygous manner. One was homozygous for a novel mutation, which was termed as "c.153-155insCT", while one of the cases was homozygous and another was compound heterozygous for the second novel mutation c.923G>A. Moreover three known mutations c.18-21delGACT, c.1017G>A, and c.681G>A in 11 of the patients were detected. No apparent mutation was observed in the rest of patients (44%, n=11).
Conclusion: The present data exhibit a fundament for molecular carrier detection and prenatal diagnosis of a relatively large percentage of Iranian patients suffering from NC, at least in the Southwestern Iran, where Arab ethnicity is one of the common ethnicities of the region.
Key words: Nephropatic cystinosis, CTNS gene, Iranian, Molecular characterization, Novel mutation.
RESUMEN
Objetivos: La cistinosis nefropática (CN) es una rara enfermedad metabólica que se debe a la mutación del gen CNTS, para que el que ya se han registrado más de 90 mutaciones diferentes. El estudio se realizó para investigar las mutaciones del gen CTNS y su promotor en un determinado número de pacientes iraníes con CN.
Métodos: Para la determinación molecular del gen CTNS en 25 pacientes con CN procedentes de 24 familias iraníes no emparentadas, se han llevado a cabo análisis con reacción en cadena de la polimerasa y con secuenciación directa.
Resultados: Ninguno de los pacientes mostró deleción de 57-kb ni heterocigócita ni homocigótica. Uno resultó homocigoto para una mutación no descrita que se denominó «c.153-155insCT», mientras que otro de los casos resultó homocigoto y otro heterocigoto compuesto para una segunda mutación no descrita, c.923G>A. Además, se detectaron otras tres mutaciones conocidas c.18-21delGACT, c.1017G>A y c.681G>A en 11 pacientes. En el resto de los pacientes (44 %, n = 11) no se observaron mutaciones aparentes.
Conclusión: Los datos de este estudio muestran un fundamento para la detección de transportadores moleculares y el diagnóstico prenatal de un porcentaje relativamente elevado de pacientes iraníes que sufren CN, al menos en el sudoeste de Irán, donde la etnia árabe es una de las comunes en la región.
Palabras clave: Cistinosis nefropática, Gen CTNS , Paciente iraní, Determinación molecular, Mutación no descrita.
Introduction
Cystinosis is a rare autosomal recessive metabolic disorder with an incidence rate of 1 per 100,000-200,000 of the world population.1 No official statistics has been released on the incidence of cystinosis in Iran; however, according to an extrapolated statistics, a frequency of 1 per 130,000 has been estimated in the Middle East. The disease is characterized by increasing levels of intracellular cystine in leucocytes and first described by Schneider et al, in 1960s, as a lysosomal storage disorder.2
Cystinosis is classified into three clinical forms according to the age of onset and severity. The most severe form is called 'infantile' or 'nephropatic'cystinosis, which results in poor growth and kidney complications in infants, known as renal Fanconi syndrome.3 The two less severe variants are 'intermediate' or 'juvenile' form with milder renal manifestations and 'ocular' or 'adult' form without affecting the kidneys.4
Nephropatic form patients are usually asymptomatic at the time of birth, but they demonstrate symptoms such as acidosis, vomiting, dehydration, hypophosphatemic rickets, and poor growth at the age of 6-12 months.1,3 Administration of cysteamine in form of oral medication or eye drops, which helps to deplete cystine from lysosomes, along with renal transplantation has a dramatic effect on ameliorating the disease.5
The three types of cystinosis are actually caused by the accumulation of crystal form cystine in lysosomes and impaired cystine export from lysosome to cytosol as a result of the absence or dysfunction in cystine transporter protein, the cystinosin. Cystinosin is a protein with 367 amino acid and 7 transmembrane domains, which is primarily expressed in lysosomal membrane.6,7 This protein is encoded by theCTNSgene locating on the 17p13.8
Some available evidences point to the CTNS as the only gene, which is responsible for clinical manifestations of the disease. To date, more than 90 various mutations have been described in different parts of the CTNS gene including exons, splicing sites and the gene promoter.9,10 It is probable that some disease-causing mutations occur more frequently in certain geographic regions. For instance, a 57kb deletion has been reported as the most common mutation in northern parts of Europe.10 Moreover, a relatively large set of pathogenic point mutations, deletions, and insertions have been defined as the molecular genetics cause of the disease,9 however, little is known about the mutation spectrum of theCTNSgene in the Middle East, especially in Iran. The only accessible data of Iranian NC patients are three 'clinical' reports.11-13
Accordingly, the aim of this investigation was to screen the mutations ofCTNSgene and its promoter in a number of Iranian patients with Nephropatic Cystinosis.
Methodology
Patient's history
Twenty-five individuals (14 males and 11 females) were collected from 24 unrelated families with age range of 7 month to 11 year old. The patients were from Arabian and Persian ethnic groups of Iran. We should mention here that one patient (P11) came of a family with cross-marriages between Arabian and Persian parents. Twenty of patients (80%) were from first cousin parents, three cases (12%) from second cousin parents, and two (8%) from a non-consanguineous marriage. Patients with the following criteria were considered as cases of cystinosis: i) signs and symptoms of Fanconi syndrome and ii) Presence of cystine crystals in cornea using slit lamp. The most common clinical features in the patients were growth retardation (100%), presence of cystine crystal in cornea (100%), rickets (88%) and polyuria-polydipsia (84%). The most frequent laboratory data were glucosuria (100%), renal tubular acidosis (96%), proteinuria (88%) and hyposthenuria (84%) (Table 1).
Table 1. Clinical findings in patients with cystinosis 
a P 7c P7 are affected sisters.
After obtaining the informed consent of all voluntary families, blood samples were collected from individuals with Nephropatic Cystinosis and their parents who were attending Abuzar Paediatrics Hospital of Jondi Shapour Medical Science University of Ahvaz (southwestern Iran).
Molecular genetic analysis
Genomic DNA was extracted from EDTA-anticoagulated samples by standard procedures. Initially, the genomic DNA of the patients were screened for the most common large 57kb deletion as has been described previously.14
The polymerase chain reaction (PCR) was carried out by a set of self- designed primers using primer3out software (www.primer3out.com) for exons 3-12. The primer sequences are available by request. The promoter region of the CTNS gene was amplified with previously reported primer pair.15 Direct sequencing of the PCR products were carried out using ABI automated sequencer 3700 in accordance with the manufacture's instruction. Sequence analysis was performed with chromas software and NCBI-Blast.
Results
GAP-PCR was carried out in all samples to detect the 57kb deletion in CTNS gene. None of the NC patients showed mentioned large deletion in homozygous or heterozygous status. It means that all of patients present the 266 bp fragment which is related to D17S829 marker; therefore the 423 bp specific fragment was not detected because this fragment is only generated in the case of deletion.
Direct sequencing of the entire gene demonstrated pathogenic mutations in 14 patients. Two patients were compound heterozygous for the c.1017G>A (leading to the G339R amino acid exchange) and the 681G>A mutation, which leads to anaberrant splice site. Nine other patients were homozygous for the 681G>A mutation. One patient was homozygous for an unreported insertion of two nucleotides (CT), which we term as 'c.153-155insCT' (Figure 1).

Figure 1. Autoradiogram
The autoradiogram indicates partial sequence of the CTNS gene presenting the novel mutation c.153-155insCT that has been shown in the affected person (upper image) and in one of the healthy parents (lower image).
Accordingly, it is obvious that parents were heterozygous for that mutation. Furthermore, two other patients showed the second novel mutation c.923G>A in this study (Figure 2).
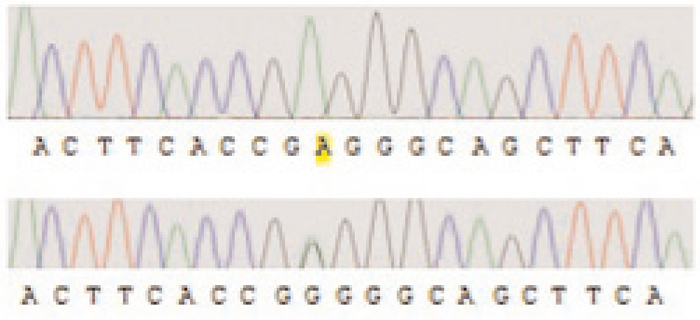
Figure 2. Autoradiogram
The second novel mutation 923G>A is illustrated in the affected person being homozygous (upper) and in one of the parents (lower) showing the mutation in heterozygous manner.
No mutation was observed in the remaining cases.11 A summary of all identified mutations were depicted in Table 2.
Table 2. Patients with CTNS gene mutation 
aP 4c is P 4's carrier brother; b P 7c & P7 are sisters; c P11c is the brother of P11; d P19c is P11's brother.ND: no data.
In addition, a number of SNPs were found in coding exons and promoter region of the CTNS gene (Table 3). Also we have identified an unreported SNP in the CTNS gene promoter in one patient and her healthy brother. Both children were heterozygous for the SNP at position -3779 (C>T) prior to the starting of nucleotide at position +1.
Table 3. Summary of all detected SNP's in the individuals being diagnosed with NC 
A total of 98 healthy non-related individuals (55 and 43 with Arabian and Persian background, respectively) were screened for the two novel mutations found in the survey. None of healthy samples were carrier of mentioned changes on genomic level.
Discussion
In current study, we investigated 25 patients from 24 unrelated families in the southwest of Iran, a region with plenty of diverse ethnic groups and mixed population, which is characterized by the vast range of consanguineous marriage. According to the previous report, the overall rate of consanguineous marriage in Iran is 38.6%, while, first cousin marriages (27.9%) is the most common form. Interestingly, significant differences were reported in prevalence and patterns of consanguinity, among ethnic/religious populations and geographical regions within the same ethnic groups.16
Initially, we searched for the 57kb deletion encompassing CTNS gene causative for fully disrupted gene function. The deletion breakpoints of this deletion extend from within exon 10 of CTNS gene and an adjacent upstream region (in CARKL/SHPK genes).17 None of investigated individuals showed the mentioned deletion. Moreover, recent reports from Saudi Arabia and our neighboring country, Turkey, confirm the lack of this deletional mutation in cystinosis patients as well.18,19 Therefore, we suggest the possibility of the absence of 57kb deletion in Middle East, at least in Iranian cystinosis patients. This large deletion seems to be specific to European population.20 It is evident that the occurrence of the 57kb deletion is associated with the most severe form of cystinosis.21,22 Recently, Freed et al. have demonstrated the extension of this deletion even into TRPV1 gene which leads to the transcription dysregulation in peripheral blood mononuclear cells isolated from cystinosis patients. It can be speculated that the involvement of TRPV1 gene may have consequences in cystinosis pathophysiology.17 Accordingly, and because of CARKL gene removing in the mutation and its impaired role in phosphorylation of sedoheptolose -that presumably will effects on the cell redox system-, we postulate that 57kb deletion positive patients may have different clinical presentation in comparison with deletion negative individuals including the present study subjects. However, a large number of cystinosis patients should be investigated in both phenotypic and genotypic assays to confirm this hypothesis.
The outstanding outcome of this study is identification of two novel mutations in CTNS gene, as well as a novel SNP in promoter of the gene. One patient and her healthy brother showed the same changes in the gene promoter. These findings lead us toward this hypothesis that mentioned SNP might have no effect on the gene expression. Further studies could clarify our supposition.
However, one of the novel detected mutations is c.153-155insCT, which creates a premature stop codon at codon 56 that leads to the production of a truncated form of cystinosin. This mutation was only observed in a 17-month-old sample with Persian background, but not in any individuals coming from Arabian families. Therefore, we speculate that the stated mutation may occur frequently in other regions of Iran with unmixed ethnicities.
The second novel mutation, c.923G>A, was detected only in two individuals with Arabian background; one was homozygous and the other one was compound heterozygous for the mutation (Table 2).
Interestingly, Kiehntopf, Shotelersuk and their colleagues have heretofore reported two missense mutations at the same codon of 308 in exon 11 of CTNS gene, in accordance with our findings.23,24 Kiehntopf reported a transversion of G>T at this codon that results in the amino acid exchange of Glycine to valine.23 Shotelersuk et al. found a missense mutation of'c.922GGG>AGG', which substitutes Glycin by Arginine.24 Here, we found a transitional mutation G>A, with the same consequence regarding the amino acid level (Figure 3). Hence, the novelty is on the altered nucleotide and amino acid, but not on the position of amino acid.

Figure 3. The amino acid and DNA sequencing of exon 11
Analogy in the number of codon; difference in the changed base and amino acid.
One patient was homozygous for the mutation c.18-21delGACT, which most likely occurs in European population.23 It is wondering that the most common mutation in present study is the 681G>A, which has recently been reported from cystinosis affected individuals of Saudi Arabia.18 Therefore, migration and/or ethnicity could be the explanation for occurrence of related mutations in Iran and Saudi Arabia, leading to the concept of possible founder mutations in Middle East. Nevertheless, this suggestion has to be verified in a larger sample size.
Furthermore, we could not find any mutation in 44% (n=11) of our subjects, this is in consistence with a number of previous mutation surveys which revealed no apparent mutation in the CTNS gene in some cystinosis patients.16,24-26 Regarding to the heterogeneous population structure in the southwestern Iran, probability of mutation diversity among Iranian cystinotic patients is expected in comparison to other populations. This diversity will be reflecting in possible existence of other forms of the CTNS gene mutations like splice site mutations in our individuals. This kind of mutation is not detectable by DNA analysis, exclusively. Therefore, it is likely that a remarkable number of cases in the present study need to be analyzed on the RNA level.
In the case of observing no mutation in patients' mRNA in future supplementary studies and considering the uncertified role of CTNS as the only gene related to the disease, it could be postulated that other unknown mechanisms might be involved in the pathogenesis of the nephropatic cystinosis at least in Iranian patients.
Nevertheless, the present data extend the mutation spectrum of the CTNS gene in NC patients and could be basically used for the prenatal or preimplatation genetic diagnosis in the region. One of the most important causes of the increased rate of rare autosomal recessive disorders like cystinosis in Iran is the relatively large number of consanguineous marriages. Therefore, to prevent and/or limit these inheritable diseases, genetic counseling before marriage, prenatal diagnosis in carriers and also in suspicious families is suggested.
Conflict of interest
The authors declare that there is no conflict of interest associated with this manuscript.
References
1. Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med 2002;347:111-21. [ Links ]
2. Schneider JA, Bradley K, Seegmiller JE. Increased cystine in leukocytes from individuals homozygous and heterozygous for cystinosis. Science 1967;157:1321-2. [ Links ]
3. Gahl WA, Thoene JG, Schneider JA. Cystinosis: a disorder of lysosomal membrane transport. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds.).The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. p. 5085-6108. [ Links ]
4. Anikster Y, Lucero C, Guo J, Huizing M, Shotelersuk V, Bernardini I, et al. Ocular nonnephropathic cystinosis: clinical, biochemical, and molecular correlations. Pediatr Res 2000;47:17-23. [ Links ]
5. Dohil R, Fidler M, Gangoiti JA, Kaskel F, Schneider JA, Barshop BA. Twice-daily cysteamine bitartrate therapy for children with cystinosis. J Pediatr 2010;156:71-75. [ Links ]
6. Cherqui S, Kalatzis V, Trugnan G, Antignac C. The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. J Biol Chem 2001;276:13314-21. [ Links ]
7. Kalatzis V, Nevo N, Cherqui S, Antignac C. Molecular pathogenesis of cystinosis: effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum Mol Genet 2004;13:1361-71. [ Links ]
8. The Cystinosis Collaborative Research Group. Linkage of the gene for cystinosis to markers on the short arm of chromosome 17. Nat Genet 1995;10:246-8. [ Links ]
9. Taranta A, Wilmer MJ, Van den Heuvel LP, Bencivenga P, Bellomo F, Levtchenko EN, et al. Analysis of CTNS gene transcripts in nephropathic cystinosis. Pediatr Nephrol 2010;25:1263-7. [ Links ]
10. Wilmer MJ, Emma F, Levtchenko EN. The pathogenesis of cystinosis: mechanisms beyond cystine accumulation. Am J Physiol Renal Physiol 2010;299:F905-F916. [ Links ]
11. Imanzadeh F, Salehpour Sh, Nariman Sh, Sayyari AA. Cystinosis Report of a Case. Iran J Pediatr 2002;13:37-41. [ Links ]
12. Mirdehghan M, Ahmadzadeh A, Bana-Behbahani M, Motlagh I, Chomali B. Infantile cystinosis. Indian Pediatr 2003;40:21-24. [ Links ]
13. Nakhaii S, Hooman N, Otukesh H. Gastrointestinal manifestations of nephropathic cystinosis in children. Iran J Kidney Dis 2009;3:218-21. [ Links ]
14. Anikster Y, Lucero C, Touchman JW, Huizing M, McDowell G, Shotelersuk V, et al. Identification and detection of the common 65-kb deletion breakpoint in the nephropathic cystinosis gene (CTNS). Mol Genet Metab 1999;66:111-6. [ Links ]
15. Mason S, Pepe G, Dall'Amico R, Tartaglia S, Casciani S, Greco M, et al. Mutational spectrum of the CTNS gene in Italy. Eur J Hum Genet 2003;11:503-8. [ Links ]
16. Saadat M, Ansari-Lari M, Farhud D. Consanguineous marriage in Iran. Ann Hum Biol 2004;31:263-9. [ Links ]
17. Freed KA, Blangero J, Howard T, Johnson MP, Curran JE, Garcia YR, et al. The 57 kb deletion in cystinosis patients extends into TRPV1 causing dysregulation of transcription in peripheral blood mononuclear cells. J Med Genet 2011;48:563-6. [ Links ]
18. Aldahmesh MA, Humeidan A, Almojalli HA, Khan AO, Rajab M, AL-Abbad AA, et al. Characterization of CTNS mutations in Arab patients with cystinosis. Ophthalmic Genet 2009;30:185-9. [ Links ]
19. Topaloglu R, Vilboux T, Coskun T, Ozaltin F, Tinloy B, Gunay-Aygun M, et al. Genetic basis of cystinosis in Turkish patients: a single-center experience. Pediatr Nephrol 2012;27:115-21. [ Links ]
20. Touchman JW, Anikster Y, Dietrich NL, Maduro VVB, McDowell G, Shotelersuk V, et al. The genomic region encompassing the nephropathic cystinosis gene (CTNS): complete sequencing of a 200-kb segment and discovery of a novel gene within the common cystinosis-causing deletion. Genome Res 2000;10:165-73. [ Links ]
21. Wilmer MJ, Levtchenko EN. Diagnosis and Treatment of Nephropathic Cystinosis. European Nephrology 2009;3(1):45-8. [ Links ]
22. Sonies BC, Almajid P, Kleta R, Bernardini I, Gahl WA. Swallowing dysfunction in 101 patients with nephropathic cystinosis: benefit of long-term cysteamine therapy. Medicine (Baltimore) 2005;84:137-146. [ Links ]
23. Kiehntopf M, Schickel J, Gönne B, Koch HG, Superti-Furga A, Steinmann B, et al. Analysis of the CTNS gene in patients of German and Swiss origin with nephropathic cystinosis. Hum Mutat 2002;20:237. [ Links ]
24. Shotelersuk V, Larson D, Anikster Y, McDowell G, Lemons R, Bernardini I, et al. CTNS mutations in an American-based population of cystinosis patients. Am J Hum Genet 1998;63:1352-62. [ Links ]
25. Alcantara-Ortigoza MA, Belmont-Martinez L, Vela-Amieva M, Gonzalez-Del A. Analysis of the CTNS gene in nephropathic cystinosis Mexican patients: report of four novel mutations and identification of a false positive 57-kb deletion genotype with LDM-2/exon 4 multiplex PCR assay. Genet Test 2008;12:409-14. [ Links ]
26. Kalatzis V, Cohen-Solal L, Cordier B, Frishberg Y, Kemper M, Nuutinen EM, et al. Identification of 14 novel CTNS mutations and characterization of seven splice site mutations associated with cystinosis. Hum Mutat 2002;20:439-46. [ Links ]
![]() Correspondence:
Correspondence:
Hamid Galehdari
Department of Genetics
Faculty of Sciences
Shahid Chamran University of Ahvaz, Ahvaz, Iran
Galehdari187@yahoo.com
Enviado a Revisar: 14 Jun. 2012
Aceptado el: 25 Sep. 2012

