










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroduction
Cystinosis, OMIM# 219800, is a lysosomal storage disease due to impaired transport of cystine across the lysosomal membrane. The gene defect involves CTNS (OMIM 606272; GenBank NM_001031681), located on chromosome 17p13,1) and results in cystine accumulation in most cells and tissues of the body, including the kidneys, eyes, bone marrow, intestines, liver, and spleen. Cystinosis is inherited in an autosomal recessive fashion; all affected individuals have biallelic CTNS mutations.
Cystinosis has been divided into three categories: nephropathic cystinosis, intermediate cystinosis and nonnephropathic or ocular cystinosis, although these subtypes actually reflect a continuum of severity. As the most common and severe phenotype, nephropathic cystinosis manifests in infancy, causing poor growth and renal Fanconi syndrome, characterized by polyuria, polydipsia, proteinuria, acidosis and urinary loss of various salts and nutrients. Renal tubular dysfunction leads to the loss of essential minerals, impairing growth and causing hypophosphatemic rickets. By one year of age, cystine crystals are generally apparent in the corneas on slit lamp examination by an experienced ophthalmologist. These crystals cause pain and increased sensitivity to light.2,3 untreated children experience complete kidney failure by approximately 10 years of age. Long-term complications include muscle deterioration, blindness, inability to swallow, diabetes, hypothyroidism, central nervous system involvement, and male infertility.
The signs of intermediate cystinosis are the same as for nephropathic cystinosis, but they typically appear in adolescence rather than in infancy. Kidney failure and corneal crystals are the most important early features of this disease. If intermediate cystinosis is left untreated, complete kidney failure can occur in the late teens to mid-1920s.3 Individuals with ocular cystinosis typically experience photophobia, but usually do not develop kidney failure or other clinical manifestations of cystinosis. Due to the absence of severe symptoms, the age at which this form of cystinosis is diagnosed varies widely.4,5
Kidney transplantation has significantly extended the lives of patients with nephropathy cystinosis. In addition, the cysteine-lowering drug, cystamine, depletes cells of cysteine by combining with cystine to produce cysteine and cysteine-cystamine mixed disulfide, which exits cystinotic lysosomes via a functional lysine carrier.6 This therapy has been salutary with respect to preservation of renal function, stimulation of growth, and prevention of the late, nonrenal complications of cystinosis.
The CTNS gene has 13 exons, but exons 3-12 are protein-coding; biallelic mutations lead to significant loss of the lysosomal cystine transporter, cystinosin.3,7-9 Approximately 200 CTNS mutations have been reported in cystinosis patients, the most common is a 57-kb deletion responsible for 50% of all the North American and Northern European cases.10-20 Here we report the CTNS mutations present in 28 Iranian patients of diverse ethnicity, providing the first molecular genetic study of cystinosis in this population.
Methods
Patients
Studies were conducted in patients, family members and normal control subjects after informed consent was obtained according to Iran University ethical committee codes. Blood samples were collected from individuals with nephropathic cystinosis and their parents who were attending Ali-Asghar Children Hospital of Iran University of Medical Science. The clinical and molecular genetics survey was performed in accordance with the Declaration of Helsinki.
During two years of study, 28 individuals (16 males and 12 females) from 28 unrelated consanguineous families participated in our studies. The patients were from different Persian ethnic groups of Iran, and inclusion required signs and symptoms of Fanconi syndrome and/or the presence of cystine crystals in the cornea, documented by a nephrologist and/or ophthalmologist, respectively. All patients had a clinical history consistent with the disease, including kidney and/or eye involvement; patients were generally referred to our clinic for genetic counseling and molecular genetic analysis by pediatric nephrologists.
PCR amplification of CTNS
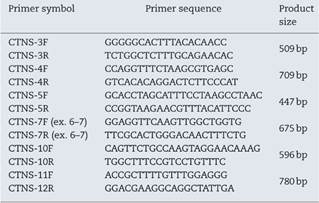
Genomic DNA was extracted as previously described.7 The 5′ UTR and the open reading frame (ORF) of CTNS (with the exon-intron boundaries) were amplified by independent PCR amplifications utilizing exon-specific primer pairs, which the sequences of some are listed in Table 1. PCR was performed in a final volume of 50 μl containing 200μM dNTP, 1 × Taq DNA polymerase buffer, various concentration of MgCl2, 2U Taq proof-reading (Pfu) DNA polymerase (Cinagen, Tehran, Iran), 200 ng genomic DNA and 10 pmol of each primer. After initial denaturation for 5 min at 95°C, 35 cycles of amplification were performed as follows: 30 s at 95°C, 30 s at 51-57°C (depends on the primer pairs) and 30 s at 72°C. PCR products were evaluated on a 1.5% agarose gel stained with RedSafe (Cinagen, Tehran, Iran).
DNA sequencing
Sequence analysis of PCR products from the promoter region and all exons was performed after purification of PCR products (PCR product purification kit, Roche). Both strands were sequenced using the Big Dye Termination system in an ABI 310 capillary sequencer (Macrogen, South Korea). Sequencing results were analyzed using bioinformatic tools (http://www.ncbi.nlm.nih.gov/gorf/bl2.html).
Results
GAP-PCR was carried out in all samples, and no patient showed the 57 kb CTNS deletion in either the homozygous or heterozygous state (Fig. 1). Direct sequencing of the entire CTNS gene demonstrated alterations in all patients. In 14 of 28 patients (50%), the alterations were observed in exons 6 and 7. Four patients (14.15%) had c.969C>A; p.N323K, in exon 11 of the CTNS gene which was previously reported, 3 were heterozygous and one was homozygous. Three patients (11%) were homozygous for a single base pair deletion in nucleic acid position 323 (c.323delA) in exon 6, leading to premature truncation of the protein (p.Q108RfsX10) (Fig. 2A). Also in exon 6, two patients (7.2%) showed homozygosity for an unreported deletion of two nucleotides, (c.257-258delCT; p.S86FfsX37). Two patients were homozygous for a novel single base pair insertion (c.92insG; p.V31GfsX28) in exon 4, and two others had a homozygous deletion (c.120delC; p.T40TfsX10) in exon 4. Another frame-shift mutation was identified as a novel homozygous single base pair deletion in exon 9 (c.661insT; p.V221CfsX6) in one patient (Fig. 2B). Also, one patient had a previously reported mutation (http://www.ncbi.nlm.nih.gov/clinvar/variation/188834/), c.18-21del GACT; p.T7fsX7, in CTNS exon 3. The variations in -303 5′ UTR of the CTNS, previously reported for ocular cystinosis,15 were detected in 2 patients in our study too.

Fig. 1 Genomic PCR amplifies CTNS amplicons. Genomic PCR amplifies the exons using the gene-specific pairs of primers. The common 57-kb deletion was not observed in any of the 28 Iranian patients. The PCR products were subjects for Sanger sequencing using relevant forward and reverse primers after purification of PCR products (PCR product purification kit, Roche).

Fig. 2 Chromatogram (Chromas software version 2.4.1) for the region containing some novel mutations. Red arrow shows the substituted nucleotide. Sequence analysis of PCR products from the promoter region and all exons was performed. Both strands were sequenced using the Big Dye Termination system in an ABI 310 capillary sequencer (Macrogen, South Korea). A, shows a single base pair deletion in nucleic acid position 323 (c.323delA) in exon 6, leading to premature truncation of the protein p.Q108RfsX10 which was observed in three patients (11%) in homozygous state. B, c.661insT; p.V221CfsX6 is a novel homozygous single base pair deletion in exon 9 observed in one patient. C, the variant c.779C>T; p.T260I (in exon 10) was detected in 18 of 28 (64%) patients. D, c.261T>A; p.F87L in exon 6 of CTNS is the most common mutation in our population which was observed in six patients (21.4%) in heterozygote and homozygote states.
In our population, the variant c.779C>T; p.T260I (in exon 10) was detected in 18 of 28 (64%) patients with nephropathic cystinosis (Fig. 2C). Furthermore, a second CTNS mutation was detected in 16 of 18 patients. The most common mutation in our population was c.261T>A; p.F87L in exon 6 of CTNS; this was observed in six patients (21.4%) (Fig. 2D).
Five patients were compound heterozygous for the mutations c.73A>T; p.S25C/c.261T>A; pF87L, c.969C>A; p.N323K/c.1015G>A; p.G339R, c.73A>T; p.S25C/c.969C>A; p.N323K, and c.261T>A; pF87L/c.969C>A; p.N323K.
A summary of all identified mutations is depicted in Table 2. In addition, a number of SNPs were found in coding exons of the CTNS gene (Table 3). A total of 68 healthy non-related Persian individuals were screened for the novel mutations found in this survey; none carried any of the changes.
Table 2 Summarized mutations detected in patients with cystinosis in Iran. All variations have been verified with the mutations reported in ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/?term=ctns%5Bgene%5D).

Table 3 Summary of CTNS mutations in cystinosis (by ClinVar) (http://www.ncbi.nlm.nih.gov/clinvar/?term=ctnsall).

Discussion
The incidence of cystinosis is estimated to be between 1 in 100,000 and 1 in 200,000 worldwide; it is predicted to be much higher in Iran due to the nation's high rate of consanguinity. The CTNS gene spans 26.6 kbp and contains 13 exons. A 57,257-bp deletion8 was found to be responsible for approximately half of Northern European and North American cystinosis patients10,11; this deletion is easily detected by a multiplex PCR amplification assay.21 Cystinosis has been reported in different ethnic groups, and about 200 different mutations have been described to date, including mutations in the promoter region, missense, nonsense, deletion, insertion, and splice-site mutations.9-11,22 However mutational spectrum of CTNS in African, Mediterranean, and middle east populations seems to be different (Table 4). Meanwhile in a population of Saudi Arabian origin, it showed small deletions and point mutations.24 None of the Egyptian patients with infantile nephropathic cystinosis who underwent molecular analysis had the 57-kb deletion,24 consistent with a study in Turkey.12 These patients showed frameshift, nonsense, missense, and intronic splicing mutations. Previous studies of nephropathic cystinosis in patients of unrelated families from southwest Iran also showed point mutations.13 Also we have not found this CTNS large deletion in any of screened Iranian patients with cystinosis. Since the 57-kb deletion originated approximately 1500 years ago in northern Europe,9 it is not surprising that it is absent among Iranian cystinosis patients.
Table 4 Summarized previous reports of genetic diagnosis of cystinosis in African and Mediterranean countries.

In this study, most of the patients showed novel mutations although some previously reported mutations have also been detected. Moreover, half of the mutations have been observed in the exons 6 and 7 of the CTNS gene, which is highly suggestive of genetic analysis of these two exons by Sanger sequencing as Tier 1 in Iranian population. Utilizing this recommendation in suspected patients with nephropathic cystinosis in Iran, the process of mutation detection is facilitated, and the cost and time of CTNS mutation detection may be reduced.
The c.779C>T was primarily described in a heterozygous genotype in 5 Italian and some other Jordanian patients.7,8 Moreover, its association with some mutant alleles has been introduced as an additional DNA marker for prenatal molecular diagnostics of cystinosis in Jordanian families.8 Also G339R, one detected mutation in a patient, has already been proposed as a common cause of nephropathic cystinosis in the south western Ontario Amish Mennonite population.20 Overall, all 28 patients in this study including the ones with novel mutations presented with Fanconi syndrome between 6 and 24 months old, and also developed eye involvement. So we admit that clinical feature of the studied patients was consistent with and similar to what is already mentioned in the literature.

