










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkSr. Director:
El síndrome de Alport (SA), descrito por primera vez en 19271, es una enfermedad hereditaria que afecta a las membranas basales. Su prevalencia estimada es de 1/5.000 a 1/10.0002. Se caracteriza por la presencia de hematuria asociada a sordera neurosensorial, lesiones oculares y alteraciones de la membrana basal glomerular que conducen, eventualmente, a insuficiencia renal crónica terminal (IRCT) y es responsable del 1-2% de los pacientes con IRCT en los países occidentales3.
En el SA la alteración se localiza en la membrana basal glomerular que está constituida por una red de colágeno tipo IV formada por trímeros de cadenas α3, α4, α5, codificadas por los genes COL4A3, COL4A4 y COL4A5, respectivamente4. El SA se transmite mayoritariamente (80-85% de los casos) mediante herencia ligada al X (SALX) con mutaciones en el gen COL4A5. Los varones están más severamente afectados, por presentar la mutación en hemicigosis, y ser las mujeres portadoras heterocigotas del gen alterado con afectación clínica generalmente más leve5. El 10-15% restante, corresponde a una herencia autosómica recesiva, con mutaciones en el gen COL4A3 o COL4A46. De forma clásica, se incluía un tercer tipo de herencia autosómica dominante, que hoy en día se engloba dentro de la llamada «nefropatía del colágeno IV, α3-α4».
Presentamos 2 casos de SA en pediatría; 2 niñas hermanas, portadoras de una mutación ligada al X en el gen COL4A5 no descrita hasta el momento.
Niña de 13 años, en seguimiento desde el año de vida por microhematuria y proteinuria no nefrótica. Como antecedentes familiares (fig. 1), la madre de 45 años presenta proteinuria y hematuria desde los 11 años con filtrado glomerular normal en la actualidad, y necesidad de audífonos desde los 37 años. La abuela materna fue diagnosticada de un síndrome nefrítico con 14 años, falleciendo en programa de hemodiálisis por nefropatía sin filiar. No existían antecedentes de enfermedad renal en la familia paterna.
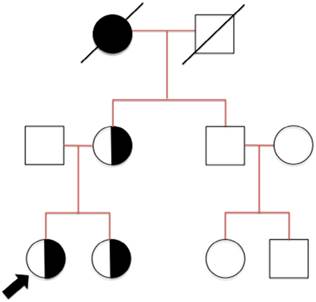
Figura 1 Antecedentes familiares. La flecha indica el caso índice. Hermana y madre portadoras a su vez de la mutación. Tío materno sano. Abuela afecta fallecida.
Debido a la clínica y a los antecedentes familiares se realizó una biopsia renal a los 5 años objetivándose en la microscopia óptica una glomeruloesclerosis segmentaria y focal. La microscopia electrónica mostró alteraciones en la membrana basal de los capilares con diferencia de espesor y rotura en paralelo, originando hendiduras con desaparición en puntos de la lámina densa. Todo ello altamente sugestivo de afectación renal en el seno de un SA. La audiometría fue normal.
Su hermana 3 años menor, comenzó a ser estudiada debido a los antecedentes familiares, objetivándose desde los 2 años proteinuria no nefrótica y microhematuria, sin alteraciones en la audición.
No se realizó biopsia renal debido a que presentaba un perfil clínico-analítico similar a su hermana. Actualmente ambas mantienen un filtrado glomerular normal y están en tratamiento con ARA II.
Se realizó un estudio genético tanto a la madre como a ambas hermanas, siendo los 3 casos portadoras heterocigóticas de la mutación COL4A5 c.412delG, p.(Gly138Valfs*17), no descrita hasta el momento; que confirmó la sospecha clínica de SALX (fig. 2). Se trata de una mutación de frameshift que conlleva el cambio de la pauta de lectura en el aminoácido glicina 138 dando lugar a la generación de un codón de terminación de traducción prematuro en la posición 184. Dicha mutación se prevé que dará lugar a una proteína de tamaño anómalo; solo 183 aminoácidos en lugar de los 1.685 que presenta la proteína normal (cadena α5 del colágeno iv, proteína codificada por el gen COL4A5). Hasta el momento esta mutación no ha sido descrita en la literatura, ni figura en las bases de datos de mutaciones en el gen COL4A5. Dado que es una mutación truncante se puede considerar definitivamente patogénica y, por lo tanto, la causa de la nefropatía en esta familia.

Figura 2 Gen COL4A5 con presencia de mutación no descrita previamente. El electroferograma muestra mutación truncante, con codón stop prematuro en posición 184, dando lugar a una proteína de tamaño anómalo (183 en lugar de 1.685 aminoácidos).
Han sido publicadas o reportadas en bases de datos mutacionales (The Human Gene Mutation Database [http://www.hgmd.cf.ac.uk/ac/index.php]; Leiden Open Variation Database [LOVD]; ARUP [http://www.arup.utah.edu/database/]) más de 1.000 mutaciones en el gen COL4A5 en pacientes y familias con SALX. Estas mutaciones incluyen grandes reordenamientos (7%), pequeñas deleciones (14%, como en el caso de nuestras pacientes) e inserciones/duplicaciones (6%), mutaciones missense (43%), mutaciones nonsense (6%) y mutaciones de splicing (23%)7,8.
El genotipo COL4A5 tiene un importante efecto predictor del curso de la enfermedad renal en los varones, condicionando la probabilidad de progresión a IRCT en la segunda década de la vida. Generalmente las mutaciones truncantes tienen un peor pronóstico con respecto a las missense (90% en grandes deleciones, nonsense, translocaciones; 70% en mutaciones de splicing; 50% en mutaciones missense)5,9. El genotipo COL4A5 tiene influencia a su vez en la sordera neurosensorial, presentando sordera a los 30 años el 90% de los varones con deleciones, mutaciones nonsense y de splicing, frente al 60% con mutaciones missense5,9. De igual forma, pacientes con mutaciones truncantes o de splicing, desarrollarán alteraciones visuales de 2 a 4 veces más que los que presenten mutaciones missense9,10.
Esta correlación genotipo-fenotipo no es tan aparente en las mujeres, debido probablemente a los fenómenos de inactivación del cromosoma X alterado. Sin embargo, es igualmente influyente ya que, por ejemplo, las mujeres con SALX y daño renal suelen presentar más frecuentemente mutaciones truncantes.
El diagnóstico genético es pues, una herramienta fundamental tanto para confirmar el diagnóstico como para conocer el mecanismo de herencia de cara a un consejo genético. La mutación genética existente puede incluso orientar sobre el curso evolutivo de la enfermedad. Consideramos el estudio genético, por tanto, esencial para un diagnóstico precoz y la instauración de tratamiento individualizado que evite el deterioro de la membrana basal glomerular y el consiguiente daño renal.

