










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkNutrición Hospitalaria
On-line version ISSN 1699-5198Print version ISSN 0212-1611
Nutr. Hosp. vol.28 n.2 Madrid Mar./Apr. 2013
https://dx.doi.org/10.3305/nh.2013.28.2.6307
REVISIÓN
La alimentación de la madre durante el embarazo condiciona el desarrollo pancreático, el estatus hormonal del feto y la concentración de biomarcadores al nacimiento de diabetes mellitus y síndrome metabólico
Maternal nutrition during pregnancy conditions the fetal pancreas development, hormonal status and diabetes mellitus and metabolic syndrome biomarkers at birth
F. J. Sánchez-Muniz1, E. Gesteiro2, M. Espárrago Rodilla2, B. Rodríguez Bernal1 y S. Bastida1
1Departamento de Nutrición y Bromatología I (Nutrición). Facultad de Farmacia. Universidad Complutense de Madrid. Madrid. España.
2Servicio de Laboratorio. Hospital de Mérida. Mérida. Badajoz. Extremadura. España.
Dirección para correspondencia
RESUMEN
El embarazo es una etapa de vital importancia, donde tienen lugar múltiples procesos hiperplásicos, hipertróficos, de adaptación metabólica y de preparación para la vida extrauterina. En esta revisión se analizan aspectos centrales de la nutrición durante el embarazo, tanto en la etapa embrionaria como fetal. Se exponen los cambios más importantes que tienen lugar en el páncreas fetal, con especial mención de la susceptibilidad de este órgano central en la homeostasis de la glucosa a enfrentarse a cambios nutricionales durante su desarrollo y maduración. Se comentan algunos estudios realizados en modelos animales y en la especie humana con especial mención del papel de la nutrición materna sobre la masa de células-β, la producción de insulina y otras hormonas y la sensibilidad a la insulina. Se detallan aspectos sobre las hipótesis del genotipo y fenotipo ahorrador, señalando que la hiponutrición causa adaptaciones metabólicas que permiten al futuro ser medrar en un ambiente de nutrientes y energía reducido. Se revisan algunos aspectos de la hipótesis de Barker y se indica que la adaptación metabólica que preconiza es un arma de doble filo en el mundo actual de abundancia que nos encontramos. Por último se revisan trabajos de nuestro grupo y de otros autores, en aspectos menos estudiados que relacionan la calidad de la dieta materna con alteraciones de marcadores de resistencia/sensibilidad a la insulina en el momento del parto. En especial se estudia el papel de la relación ácidos grasos saturados/hidratos de carbono y la de ácidos grasos omega-6/omega-3 en el marco de dietas inadecuadas bajo el punto de vista del índice de alimentación saludable o de la adherencia a la dieta mediterránea que condicionan en el neonato un perfil de resistencia a la insulina. La revisión incide además en que los hábitos nutricionales deben estar fuertemente instaurados ya en la etapa pregestacional para asegurar una buena alimentación desde las primeras semanas del embarazo, y asegurar un desarrollo fetal y en particular pancreático que posibilite una homeostasis adecuada de la glucosa durante el embarazo y en etapas posteriores de la vida evitando, o al menos frenando, el desarrollo y la instauración de enfermedades degenerativas asociadas con el síndrome metabólico y la diabetes tipo 2.
Palabras clave: Embarazo. Páncreas fetal. Placenta. Nutrición materna. Marcadores de sensibilidad/resistencia a la insulina. Neonatos.
ABSTRACT
Pregnancy is a vital period where several hyperplasic, hypertrophic processes together with metabolic adaptation and preparation for extra-uterine life take place. Present review accounts for central aspects of nutrition throughout gestation on the embryonic and fetal periods. It is centered in the major changes occurring in fetal pancreas, with special mention to the susceptibility of this main glucose homeostasis organ to support nutritional changes during maturation and development. Studies performed in animal models as human are commented considering the role of maternal nutrition on β-cell mass size, insulin and other pancreatic hormones production, and insulin sensitivity. Details of both the thrifty genotype and phenotype hypothesis are given, indicating that hypo/subnutrition causes metabolic adaptations that permit the future body to grow and develop itself in limited environmental and energetic conditions. The Barker hypothesis is considered suggesting that this metabolic hypothesis is a double-edged sword in the actual abundance World. Lastly the review, taking into account our own research and other papers, analyses less known aspects that relate maternal diet with insulin resistance/sensitivity markers at delivery. Particularly the role of the saturated fatty acid/carbohydrate and omega-6/omega-3 ratios in the frame of maternal diet is reviewed considering the quality of those diets under the Healthy Eating Index and the Adherence to Mediterranean Diet scores and the relationship with insulin resistance profile at birth. Present review ends indicating that nutritional habits should be strongly stated before gestation in order to assure a proper nutrition since the first moment of pregnancy. This will support an adequate fetal and pancreatic growth and development, and in turn, adequate glucose homeostasis during pregnancy and later in life, slowing down or preventing from degenerative diseases related with metabolic syndrome and type 2 diabetes mellitus.
Key words: Pregnancy. Fetal pancreas. Placenta. Maternal nutrition. Insulin sensitivity/resistance biomarkers. Neonates.
Abreviaturas
ADM: Adherencia a dieta mediterránea.
AGA: Adecuado para su edad gestacional.
AGP: Ácidos grasos poliinsaturados.
AGS: Ácidos grasos saturados.
Apo: Apolipoproteína.
DG: Diabetes gestacional.
HOMA-IR: Marcador de resistencia a la insulina en la homeostasis de la glucosa.
IAS: Índice de alimentación saludable:
IGFs: Factores de crecimiento similares a la insulina.
IMC: Índice de masa corporal.
IP: Índice ponderal.
ISR-1: Sustrato 1 del receptor de la insulina.
LGA: Grandes para su edad gestacional.
LPL: Lipoprotein lipasa.
PPLIN: Gen de la perilipina.
QUICKI: Índice de sensibilidad a la insulina.
SGA: Pequeños para su edad gestacional.
SEN: Sociedad Española de Nutrición.
GH: Hormona de crecimiento.
SNP: Polimorfismo de un solo nucleótido.
T2DM: Diabetes mellitus tipo 2.
VLDL: Lipoproteínas de muy baja densidad.
Introducción
Los problemas relacionados con el crecimiento, desarrollo y maduración constituyen actualmente un tema de gran importancia tanto bajo el punto de vista sanitario como de investigación para pediatras, endocrinólogos, nutricionistas y personal sanitario en general1,2. El fenómeno biológico del crecimiento está ligado a la multiplicación celular y por consiguiente al incremento de la masa de un tejido y por extensión del propio cuerpo. El término desarrollo hace referencia al grado de organización y complejidad funcional que alcanzan las diferentes estructuras orgánicas. Por último, maduración se refiere al nivel de desarrollo alcanzado por un tejido o por un organismo en un determinado momento3.
El embarazo es un periodo de vital importancia, tremendamente complejo, donde confluyen de forma armónica los tres conceptos previos, donde el futuro ser, además de incrementar la masa celular, se desarrolla y madura morfológicamente para adquirir de forma progresiva capacidades funcionales. En el embarazo confluyen multitud de factores que implican que la vida de un futuro ser fracase o se desarrolle con total o parcial éxito.
Desde la fecundación, acontecen en el nuevo ser multitud de procesos que incrementan paulatinamente el número y tamaño de sus células, así como su especialización funcional que le permitirán adaptarse y sobrevivir extrauterinamente. Desde el óvulo fecundado, hasta el momento del parto tiene lugar la puesta en marcha de un programa de información "impreso" en sus propios genes e influido por el estatus materno4,5; donde no sólo existen factores genéticos, sino ambientales, como la alimentación, que ejercen un papel central, sin olvidar los niveles hormonales materno-fetales y placentarios, el posible estrés, la presencia de factores tóxicos (p.e. alcohol, tabaco, fármacos), las infecciones víricas o bacterianas, etc. (fig. 1).

El conocimiento de las enfermedades hereditarias o las debidas a aberraciones cromosómicas tempranas era hasta unos años un aspecto central en la investigación perinatal y en el conocimiento de la salud del neonato. Las hipótesis de Barker6,7 o la de Palinski y Napoli8,9, entre otras, han supuesto la puesta en marcha de multitud de estudios que contemplan la interacción madre-hijo en sus múltiples facetas y la necesidad de conocer los muchos factores que regulan el desarrollo óptimo, ajustado o problemático del nuevo ser4,6-9.
En el embarazo podemos distinguir dos periodos fundamentales, el embrionario que se extiende durante las primeras 8 semanas y el fetal que abarca desde la semana 9 hasta el parto10,11 (fig. 2). Se considera que un neonato es pre-término cuando su gestación dura menos de 37 semanas, a término cuando el parto acontece entre las semanas 37 y la 42, y post-término cuando la gestación dura 42 o más semanas12-14. También otros aspectos son interesantes a tener en cuenta, ya que un recién nacido es de bajo peso cuando al momento de nacer su peso es menor de 2,5 kg, normopeso cuando se encuentra entre 2,5 y 3,999 kg y de alto peso cuando tiene más de 4,0 kg. En la actualidad se considera determinante el concepto de pequeño para su edad gestacional o SGA (acrónimo de la terminología inglesa small-for-gestational-age), apropiado para su edad gestacional (AGA, apropriate-for-gestational-age) y grandes para su edad gestacional (LGA, large-for-gestational-age)12-14 que discrimina el grado de maduración y desarrollo mejor que la propia edad gestacional.

Como puede observarse en la figura 2, las dos primeras semanas del periodo embrionario son tremendamente críticas y hacen al embrión muy vulnerable, aconteciendo, cuando las condiciones son adversas (p.e. presencia alterada de hormonas, bajos niveles de riego y nutrientes uterinos), la muerte prenatal. Este periodo de "fracaso" es conocido por el vulgo como periodo de "huevo huero" en la idea de "huevo sin embrión", en ocasiones, se acompaña de hemorragias leves a severas. En el periodo entre las semanas 2-8 el embrión es extremadamente sensible o vulnerable a los agentes teratógenos, pues coincide con la formación de los esbozos de órganos. En él pueden acontecer graves malformaciones11,15,16. Por último en el periodo fetal, pueden tener lugar alteraciones morfológicas menores, pero importantes adaptaciones fisiológicas. Esta etapa es susceptible de ajustes que dan origen a la programación fetal de la que luego hablaremos y que condicionará el desarrollo óptimo o problemático del nuevo ser11,15,16.
De acuerdo con Herrera et al.17,18 durante el embarazo, en la futura madre, acontecen dos periodos fundamentales. El primero de índole anabólico o de creación de reserva seguido de una mal llamada etapa catabólica en la que tienen lugar la movilización de las reservas creadas y la adaptación para el parto. Ambas etapas se distribuyen de manera similar en diferentes especies. Así, la primera etapa se extiende durante aproximadamente 2/3 del embarazo (p.e. 26-27 semanas en los humanos y 14 días en la rata). Este periodo anabólico coincide con incrementos marcados por parte materna de la concentración de insulina y de la sensibilidad a esta hormona, del tamaño de la placenta, del volumen de líquido amniótico, de la "reserva proteica" y del depósito de grasa en tejido adiposo de la madre, habiendo alcanzado el feto, sin embargo, un tamaño bastante reducido en comparación con la ganancia de peso materno. En la fase de distribución de reservas sigue el crecimiento lineal de la placenta y del líquido amniótico, tiene lugar un crecimiento fetal exponencial, pero se reduce enormemente el ritmo de ganancia de las reservas maternas.
En términos didácticos la madre debe ganar entre 11,3 y 15,8 kg para asegurar la gestación de un nuevo ser cuyo peso al nacimiento se encuentre entre 2,5 y 4,0 kg19. No obstante, en la actualidad, y considerando que tanto la gestación de niños SGA como la de LGA presenta graves problemas para el desarrollo futuro de enfermedades del adulto5-9,20,21 y que la ganancia excesiva o reducida también implica riegos para la madre20,21, se aconseja un incremento de peso durante el embarazo acorde con el exceso, defecto o normalidad del peso en el momento de la fecundación (peso pregestacional). Cuando existe exceso ponderal parece adecuada una ganancia entre 6,8 y 11,3 kg, mientras que en caso de madres con peso reducido, se recomienda incrementos de peso aproximados entre 12,7 y 18 kg19; lógicamente tales ganancias ponderales durante gestación serán mayores en el caso de gestación gemelar o múltiple donde serán del orden de 18 kg y matizable dependiendo del peso pregestacional y del número de fetos engendrados22. Otro aspecto importante resulta el ritmo de ganancia de peso durante el embarazo. Un ritmo apropiado podría ser de 4 kg durante las primeras 20 semanas e incrementos ponderales de 0,4 a 0,5 kg/semana en el resto del embarazo19,22.
Al final de la gestación tienen lugar "ajustes" para permitir la transición desde el seno materno a la lactancia pasando por el parto. Es una situación que implica en la madre cambios en la concentración de sustratos (p.e. lípidos plasmáticos, glucosa) cambios en la concentración hormonal (insulina, somatotropina coriónica, progesterona, prolactina) y en donde es destacable cierto grado de resistencia a la insulina. Esta situación deriva de la importancia de asegurar glucosa al cerebro materno y al feto y de dirigir glucosa a la glándula mamaria reduciendo la utilización de glucosa por otros territorios maternos17,18,20. Sin embargo, cuando la homeostasis de la glucosa en gestación no es fisiológicamente correcta, pueden sobrevenir cambios23, que en muchos casos atañen de forma grave al desarrollo del feto y lo predisponen a diabetes en la edad adulta20. En algunas mujeres no diabéticas, a lo largo del embarazo se desarrolla una alteración del metabolismo de los hidratos de carbono, sucediendo que aunque en ayunas los niveles de glucosa e insulina son normales o incluso reducidos, tras una carga de hidratos de carbono en ayunas la glucemia se eleva por encima de valores normales. Estos cambios son más acentuados en el último tercio de la gestación y se incluyen dentro del término diabetes gestacional (DG)23.
La DG se define como cualquier nivel de intolerancia a la glucosa que comienza o se diagnostica por primera vez durante el embarazo. Suele ser una condición transitoria en la que una embarazada no es capaz de producir suficiente insulina o se vuelve resistente a ésta, lo que provoca unos niveles elevados de glucemia23. La DG suele diagnosticarse en el segundo trimestre de la gestación, cuando la producción de hormonas antagonistas a la insulina (p.e. somatotropina coriónica) es mayor17,18,20. Estas hormonas modifican la sensibilidad de los receptores periféricos a la insulina, desviándose la glucosa para el feto y elevándose la movilización de los lípidos del tejido adiposo materno.
La DG ha sido ampliamente evaluada en roedores24, con resultados bastante extrapolables al ser humano. En general, ratas gestadas por madres con DG moderada secretan menos insulina y su tolerancia a la glucosa disminuye, mientras en casos de diabetes severa, se induce en las crías resistencia a la insulina en músculo esquelético e hígado. Así, los descendientes adultos de ratas diabéticas muestran una masa de células-β, una insulinemia y glucemia dentro de intervalos de normalidad, pero la respuesta insulínica a glucosa está alterada tanto in vivo como in vitro. La DG no tratada/ no compensada somete al embrión y al feto a hiperglucemia, afectándose los tejidos fetales y particularmente el crecimiento, desarrollo y maduración del páncreas fetal25,26.
Es decir, las madres diabéticas transmiten a su descendencia una tendencia diabetogénica, que no se manifiesta en situaciones normales, pero sí en aquellas situaciones donde se requiere un incremento de la metabolización de la glucosa. Es más, la obesidad y otros factores que promueven resistencia a la insulina parecen incrementar el riego de Diabetes mellitus tipo 2 (T2DM) después de DG21,27, mientras que la prole de mujeres con DG tiene incrementado el riesgo de obesidad, intolerancia a la glucosa y diabetes al final de la adolescencia y en épocas tempranas del estado adulto28. La gestación en una mujer diabética no sólo se asocia con riesgo materno, sino que también se relaciona significativamente con prematuridad, crecimiento excesivo intrauterino, síndrome de distrés respiratorio y defectos congénitos que conducen a un aumento de la mortalidad neonatal.
La DG moderada da lugar a niños con hiperplasia e hipertrofia pancreática y mayor peso26, lo que se acompaña de elevación de la insulinemia fetal. En situación severa, se induce hipertrofia con elevación temprana de los niveles de insulina, pero con estímulo permanente y excesivo del páncreas, afectando a su funcionamiento y conduciendo a hipoglucemia, disminución del metabolismo y microsomía en el periodo perinatal29.
Por ello es fundamental controlar la glucemia y la tolerancia a la glucosa durante el embarazo. Durante las semanas 24-28 de la gestación es preceptivo en la actualidad la realización de una prueba de tolerancia a la glucosa llamada test de O'Sullivan30. Así, la prueba consiste en la administración vía oral de 50 g de glucosa y medida de la glucemia 1 hora después. Cuando los niveles de glucosa después del test superan los 140 mg/dL (0,78 mmol/L) se procede a administrar una carga oral de 100 g y medir las variaciones de la glucemia cada hora durante un total de tres horas. La curva de glucemia normal en gestación supone valores máximos en ayuno, a la hora, a los 2 horas y a las 3 horas de 105; 190; 165 y 145 mg/dL, respectivamente (5,83; 10,56; 9,17 y 8,06 mmol/L, respectivamente). Cuando al menos en dos medidas se igualan o superan dichas cifras de glucemia, se diagnostica DG23. Este problema metabólico se presenta aproximadamente en el 7% de todas las embarazadas. En un estudio nuestro realizado en unas 176 gestantes se encontró que aproximadamente el 5% de ellas fue diagnosticado de DG31. De todo lo señalado se deduce por tanto que es importantísimo el tratamiento nutricional, o en su caso, farmacológico para corregir la DG. En algunos casos, la situación de DG se corrige con dietas equilibradas donde se controla el balance energético y el incremento de peso, ya que como es sabido la obesidad induce resistencia a la insulina32.
La nutrición en el embarazo
Tres son las fases nutricionales del futuro ser: Histiotrófica, histiotrófica-placentaria o mixta y placentaria (fig. 2). La fase histiotrófica (de histio, tejido, y trofos, lugar) implica que el embrión se alimenta directamente de los nutrientes y sustratos disponibles en el útero materno. Hasta el día 18 del embarazo no es manifiesto el esbozo de la circulación materno-fetal33 y por tanto de la futura placenta, por lo que el estatus nutricional de la futura madre durante las primeras semanas de vida intraútero es muy similar al estatus preconcepcional, y marcará la viabilidad del futuro ser y el comienzo correcto de la formación y estructuración de los órganos y tejidos.
Ya al inicio de la gestación (día 6), tras la implantación ha comenzado en el blastocisto la especialización de líneas celulares. Se observa el ectoblasto que originará la piel y el tejido nervioso y en entoblasto que dará lugar a las glándulas digestivas, epitelio digestivo y epitelio respiratorio, mientras que el mesoblasto, que originará el esqueleto, los músculos, el sistema conjuntivo, el aparato circulatorio y el aparato urogenital aun no es evidente16,33. Un cluster de células, el sincitio-trofoblasto comienza la invasión del útero materno con la producción de factores angiogénicos y factores de transcripción que originarán entre otras cosas la formación de la placenta y de la circulación útero-placentaria34 (fig. 3).

En interesante comentar que la placenta ha madurado como órgano central en la gestación para realizar procesos de síntesis, transferencia y modificación de sustratos una vez cumplido el primer trimestre del embarazo en los humanos20,21,33,35. Durante estas doce primeras semanas, la placenta va formándose, especializándose y se constituye como una entidad esencial en esta etapa del embarazo16,33. No obstante esta placenta es inmadura y el feto está claramente rodeado de líquido almacenado en el espacio celómico y en el espacio amniótico. El saco vitelino también se constituye como un reservorio esencial de nutrientes maternos. Estos líquidos son ultrafiltrados del plasma y aportan a través de la piel y las mucosas cantidades muy importantes de sustratos, mientras que el acceso por el futuro cordón umbilical todavía es deficitario36. Es claramente una etapa histiotrófica-placentaria (fig. 2). Durante el segundo y tercer trimestre del embarazo, madura la placenta y tienen lugar profundos cambios en ella y en el feto. La cara posterior y el saco vitelino involucionan, la placenta se especializa y la transferencia de sustratos vía cordón umbilical se hace preferente16,33,35.
Aunque el feto está especializado preferentemente para utilizar glucosa37, como lo demuestra la génesis de multitud de factores de transcripción que implican niveles elevadísimos de transportadores tipo GLUT-1, y GLUT-4, pero también de GLUT-2, GLUT-338,39, otros factores son indispensables para el buen desarrollo de la placenta y por tanto del nuevo ser. Podemos decir que durante la gestación la esencialidad de la glucosa, proteínas, ácidos grasos poliinsaturados (AGP) de las familias omega-6 y omega-3, minerales, vitaminas, etc, es indudable20-22. La importancia de la concentración, disponibilidad y transferencia del yodo ha sido internacionalmente reconocida20-22,35. Larqué et al.40,41 han revisado recientemente el papel de la placenta en la transferencia de sustratos, particularmente de AGP de la familia omega-3, y más en particular del ácido docosahexaenoico. Es indudable que la placenta al igual que otros tejidos dispone del equipo enzimático para obtener dichos ácidos grasos de los triglicéridos de las lipoproteínas de muy baja densidad (VLDL) maternas (p.e. lipoprotein lipasa)17,18,42 y que modificaciones genéticas, epigenéticas y nutricionales condicionarán no sólo el nivel y composición de las VLDL maternas, sino también la concentración y actividad de dichos enzimas y, por ende, la transferencia placentaria17,18,40,41. También recientemente se ha señalado el papel central de la vitamina D en la formación y maduración del feto y la placenta43,44. La vitamina D promueve la maduración celular e induce tanto la diferenciación como la apoptosis en diferentes líneas celulares. Algunas células del cuerpo expresan la hidroxilasa 1-α para la 25(OH)-vitamina D3, lo que sugiere pueden producir calcitriol localmente con el fin de regular su diferenciación y proliferación45.
La nutrición en el embarazo, no sólo debe mirarse bajo un punto de vista pasivo de aportar nutrientes y otras sustancias que aseguren recambio de estructuras y crecimiento, sino de un proceso donde las funciones energéticas, estructurales y de control y regulación metabólicas se deban a la interacción de los nutrientes sobre nuestros genes, aspecto que posibilitará la expresión génica y la formación de "entidades" bioquímicas (p.e receptores, transportadores, enzimas, hormonas) que hagan posible la mejor funcionalidad de los nutrientes46-50. Incidimos por tanto en el campo de la nutrigenómica, donde nuestro fenotipo será la resultante de la interacción de los nutrientes sobre nuestros genes46-50. Tampoco debemos olvidar que en virtud de mutaciones en nuestros genes, muchas de ellas silentes, la respuesta a la dieta, y por tanto la interacción de la dieta sobre nuestros genes, se modifica. Es decir, la nutrigenómica viene modulada por la nutrigenética, llevándonos al campo de las dietas individualizadas y óptimas46-50.
Aspectos generales de la alimentación durante el embarazo
Aunque un informe detallado de este aspecto se sale del contenido y objetivo de esta revisión, parece lógico recordar algunos detalles de las recomendaciones de energía y nutrientes durante la gestación51-56, así como mostrar un resumen de los objetivos nutricionales y guías de alimentación señalados más recientemente por las autoridades y Sociedades de la Nutrición19-21,52-56. En la tabla I se presenta un esquema de aquellos aspectos dietéticos de interés durante gestación. Por ello se aconseja no comer por dos, aunque sí incrementar ligeramente el tamaño de raciones y elevar el número de tomas a lo largo del día, aconsejándose 5 ó 6, para así evitar el volumen excesivo que incrementaría el riesgo de vómitos y pirosis, como una menor demanda de insulina con fines metabólicos. La última toma, la postcena o resopón parece altamente aconsejable, ya que evita el bache hipoglucémico inducido por la demanda fetal entre la cena y el desayuno, asegurando una buena utilización de glucosa para el feto y una protección de las "reservas" proteica y grasa17. Esto parece recomendable especialmente en aquellas gestantes que cenen temprano.


También debe evitarse el consumo de alimentos crudos, por el riesgo alimentario (p.e. toxoplasmoxis) que implica57. Parece importante no introducir cambios bruscos en la alimentación materna, o aconsejar alimentos que no gusten o que puedan ser extraños a la futura madre. No es la gestación momento para ensayos. La alimentación será a la vez, variada, adecuada, equilibrada y simultánea. Es decir en cada comida deben encontrarse platos que aporten variedades de hidratos de carbono, proteínas, lípidos y micronutrientes. Una ingesta adecuada asegurará complementaciones y sinergias de nutrientes, mejorará la salud digestiva de la madre y de su microbiota y aportará componentes bioactivos imprescindibles. Conviene considerar que el consumo de alcohol, incluso de bebidas de baja graduación debe restringirse al máximo para evitar los problemas fetales relacionados con el consumo de alcohol en gestación (Síndrome alcohólico fetal o Efectos del alcohol en el feto)58-60. Hay que recordar los aspectos de malnutrición primaria o secundaria ligadas a la ingesta de alcohol y al bloqueo que el alcohol realiza sobre funciones metabólicas, más aún considerando que la placenta carece de actividad alcohol dehidrogenasa58.
Un aspecto de indiscutible interés se deriva de la recomendación de reducir el consumo de ciertos pescados grasos durante la gestación. Así, en 2011, la Agencia Española de Seguridad Alimentaria y Nutrición (AESAN) haciendo eco de lo comunicado por la Agencia Europea para la Seguridad Alimentaria (EFSA) en 200461 señaló que el consumo de atún rojo, pez espada, tiburón y lucio debía suprimirse en gestación debido al alto contenido de mercurio de estos pescados. Desafortunadamente la prensa recogió erróneamente en un comunicado dicha noticia, señalando que no debía consumirse ningún pescado graso o azul durante el embarazo. Este aspecto es de crucial importancia pues los pecados azules contienen ácidos grasos de la familia omega-3 que no se encuentran en otros alimentos. La reducción drástica del consumo de estos alimentos marinos durante gestación limitaría claramente, no sólo el aporte de tales ácidos grasos preformados al feto, sino también de la vitamina (hormona) D, con los consiguientes aspectos negativos para la madre y para el crecimiento, desarrollo y maduración del feto43,44,62-66. Desconocemos el alcance futuro de este "consejo" pero una caída del consumo de pescado graso en España, y en particular durante el embarazo, repercutiría negativamente sobre la concentración sanguínea y tisular de AGP omega-3 y por tanto sobre el cociente omega-6/omega-3, de indudable importancia en muchos aspectos fisilógicos62,64,65,67.
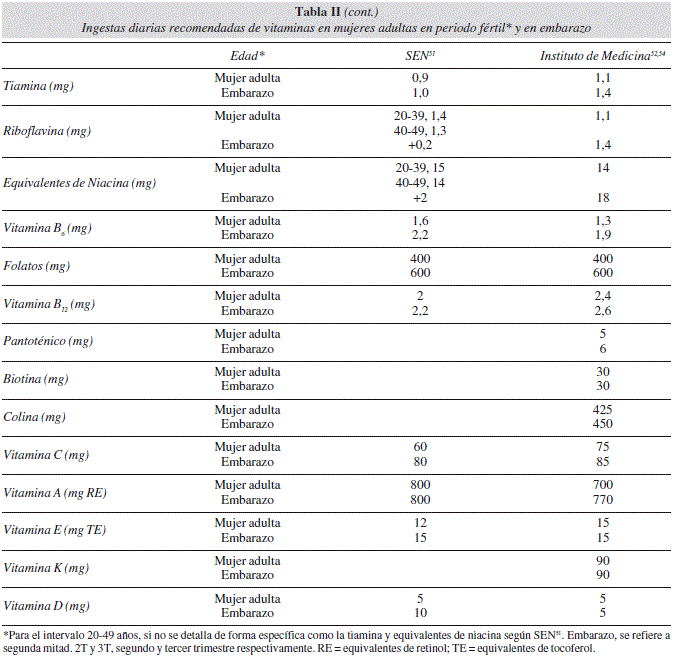
También se adjuntan las recomendaciones de energía y nutrientes tanto pregestacionales como para la segunda mitad de la gestación para mujeres adultas o entre el intervalo 20-49 años de edad (tabla II). Así, observamos que en la segunda mitad de la gestación deben incrementarse la ingesta energética [250 kcal según la Sociedad Española de Nutrición (SEN)]51, o entre 340 y 452 kcal según el Institute of Medicine americano52, aproximadamente entre un 8-15% la ingesta energética de la situación pregestacional. Proteínas y otros nutrientes también deben incrementarse en porcentajes que dependen del nutriente, pero que en algunos casos puede superar el 50%, debido a las demandas maternas y de los productos de la concepción. En la actualidad existe controversia en incrementar las recomendaciones de la vitamina A y del Fe. El primero por considerar los riesgos teratogénicos derivados del exceso del consumo de dicha vitamina (p.e incremento de la prevalencia de espina bífida)19-21,68. El segundo, por las molestias gastrointestinales y riesgo de macrocitosis69 que origina y por la competencia del Fe en la absorción de otros minerales como el Zn70.

Entre los obstetras y autoridades en el campo de la nutrición existe el acuerdo general de prescribir durante el embarazo suplementos de hierro, folatos e yodo, considerando las características pregestacionales de la población femenina. En 1986 la Organización Mundial de la Salud (OMS)71 declaraba: "La deficiencia de yodo es, mundialmente y después la inanición extrema, la causa nutricional más frecuente de retraso mental prevenible". Posteriormente, como señalan Monreale de Escobar y Obregón35 se ha incluido entre los Derechos de la Infancia de tal forma que se puede proclamar: 1- "Todo niño(a) tiene derecho a un aporte adecuado de yodo para asegurar su desarrollo normal". 2- "Toda madre tiene el derecho a una ingesta adecuada de yodo durante el embarazo, para asegurar que sus hijos(as) alcancen el desarrollo mental potencial óptimo". Estas recomendaciones implican la urgencia de consumir, incluso antes de la gestación, suplementos de yodo para incrementar las reservas tiroideas72, sobretodo en zonas donde el aporte de dicho mineral sea muy bajo, lógicamente evitando la iatrogenia indiscriminada de yodo que podría conducir a tirotoxicosis.
A su vez, se aconsejan suplementos de folatos dada la dificultad de mantener niveles de ingesta adecuada de esta vitamina (600 μg/día)51 y la relación entre hipofolatemia materna durante el primer mes de embarazo y riesgo incrementado de malformaciones del tubo neural73-76, y posteriormente con anemias macrocíticas e hiperhomocisteinemia73-76. El tema del Fe es controvertido ya que recuperar los niveles de hemoglobina y la reserva de Fe requiere tiempo y no son pocos los problemas relacionados con el exceso de hierro en el feto (p.e. hemorragias, problemas de crecimiento)69. Por eso la SEN51 (tabla II) no incrementa en sus recomendaciones las cantidades de hierro durante la gestación, ya que considera que la absorción de hierro durante la gestación se eleva del orden de 2,5 veces respecto a la situación pregestacional y que no existen pérdidas uterinas menstruales. No obstante otras entidades52,53,55, considerando que las reservas de hierro están reducidas ya pregestacionalmente y que difícilmente se adecúan a 18 mg/día durante la gestación, elevan las ingestas dietéticas de referencia a 27 mg/día para dicho mineral (tabla II), lejos aún del valor de 45 mg/día de las ingestas máximas tolerables (UL) para dicho mineral53 (tabla III).

Otro tema de interés es aconsejar que se evite la toma indiscriminada de vitaminas durante el embarazo, ya que el exceso de una puede puede interferir en las otras, ya que en muchos casos todas deben estar biodisponibles de forma simultánea. Debemos recordar la interacción entre sí, de las vitaminas B1, B2 y Niacina participando en el metabolismo de los macronutrientes, o de los folatos con las vitamina B6 y B12 en la transferencia de grupos activos, Particularmente debe evitarse el exceso de 3 de ellas68,77-79. El exceso de vitamina A, ya comentado, es teratogénico (afecta al crecimiento y estructura facial, a la disminución del crecimiento y eleva el riesgo de espina bífida)68, por ello para cubrir las ingesta recomendadas o reducir el riesgo de teratogenicidad debido al exceso de vitamina A de origen animal, debe potenciarse el consumo de β-carotenos. El exceso de vitamina B6 induce disfunción del sistema nervioso77. Por su parte un exceso de vitamina C (> 2 g/ día) eleva el riesgo de escorbuto agudo después del nacimiento, por haberse adaptado el feto a dosis altas de vitamina C69, incrementa el riesgo de cálculos renales78, interfiere con el estrógeno placentario y probablemente con el metabolismo y absorción de vitamina B12, produciendo cefaleas, fatiga, hemólisis, náuseas, vómitos, hipoglucemia e hipercolesterolemia79.
Entre los patrones dietéticos más aconsejables se encuentra el de la dieta mediterránea, basado en una ingesta elevada de productos vegetales, ricos en hidratos de carbono, fibra, compuestos bioactivos, aceite de oliva y moderado de pescado80. La dieta mediterránea es la forma de alimentación que, desde hace varios siglos, han seguido los habitantes de las regiones bañadas por el mar Mediterráneo. Aunque no existe una definición totalmente aceptada de dieta mediterránea, ésta se reconoce como el patrón de alimentación tradicional que caracterizaba a los países de la cuenca del Mediterráneo hacia la mitad del siglo XX81. Los rasgos más característicos de este patrón dietético son:
1) Alto consumo de pan, cereales integrales, frutos secos y miel.
2) Alto consumo de legumbres, verduras, hortalizas (ensaladas) y frutas frescas.
3) Grasa culinaria: aceite de oliva (cocinar y aderezo). Cocinado: Fritura.
4) Baja ingesta de grasas saturadas (cremas, mantequilla y margarina).
5) Frecuente consumo de pescado.
6) Moderada ingesta de productos lácteos: principalmente queso y yogurt.
7) Bajo consumo de carnes y productos cárnicos: embutidos curados.
8) Consumo moderado de vino.
9) Un alto consumo de ajo, perejil, cebolla, albahaca, orégano, cúrcuma, anís, sésamo, canela y otras especias.
La dieta mediterránea forma parte de un estilo de vida que engloba tanto el uso de una variedad de ingredientes tradicionales como una elaboración de las comidas de forma característica. La combinación de sus elementos dio como resultado una dieta saludable82. Aunque existen variaciones en el consumo de los componentes de la dieta mediterránea tradicional entre los distintos países del Mediterráneo82, debido a los recursos alimentarios de cada país, todos se acercan a este patrón común83. La consideración de la dieta mediterránea como dieta saludable surgió a partir de estudios nutricionales realizados en Creta, donde se detectó una incidencia de arteriosclerosis, enfermedades cardiovasculares (ECV) y enfermedades degenerativas inferior a la media de otras poblaciones y una mayor esperanza de vida80. En la tabla IV se recoge información para valorar la adherencia a la dieta mediterránea (ADM). El primer "score" fue acuñado por Trichopoulou et al.84,85, aunque posteriormente se han definido otros índices, pero en todos los casos siempre se encuentran, con matices, las mismas premisas.

PREDIMED, un estudio epidemiológico a gran escala ha utilizado un índice de ADM de 14 puntos, definido en la tabla IV y basado en las publicaciones de Estruch et al.86 y Salas-Salvadó et al.87 para conocer la relación entre obesidad y seguimiento de un patrón alimentario característico de la dieta mediterránea. Nuestro grupo ha utilizado este patrón para valorar la ADM durante gestación, eliminando la puntuación por el consumo de vino por considerar que no deben ingerirse bebidas alcohólicas durante este periodo, aunque sean de mediana o baja graduación. Se definió que la ADM era baja cuando del total de esos 13 puntos no se alcanzó una valoración de 7 puntos88.
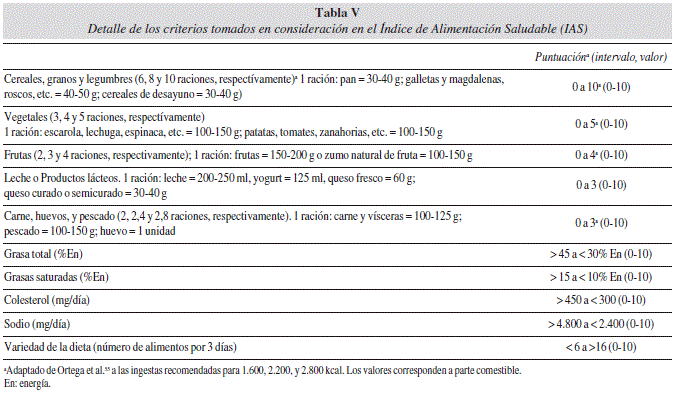
También el Índice de Alimentación Saludable (IAS) se ha empleado para la valoración de la calidad global de la dieta. Dicho índice evalúa 10 componentes de la dieta en una escala de 100 puntos y refleja la complejidad del perfil de la misma89 (tabla V). La validez del IAS se ha demostrado en estudios utilizando biomarcadores plasmáticos90,91. Sin embargo, según Koning et al.92 el IAS no predice el riesgo de T2DM en hombres. En dicho índice existen 4 apartados que valoran en la dieta el contenido total de grasa, de AGS, de sal y colesterol y otros 6 apartados que miden el consumo de grupos de alimentos y su ingesta junto con la variedad de la dieta (tabla V). El IAS ha sido utilizado y adaptado por diferentes autores para conocer la calidad de la dieta durante gestación93,94. Nuestro grupo ha empleado dicho índice para valorar la calidad de la dieta durante el embarazo, considerando que una dieta es inadecuada o de baja calidad cuando su valoración da cifras menores a 7088.
Formación del páncreas durante el embarazo
El páncreas es un órgano exocrino y endocrino fundamental para nuestra economía. Mediante la producción de enzimas contribuye de manera decisiva a la digestión de los macronutrientes95,96 mientras que el segmento endocrino es clave para la homeostasis de la glucosa97,98. De hecho, la T2DM es una patología que por su prevalencia y consecuencias fisiopatológicas a medio y largo plazo constituye un problema sanitario de primer orden99.
Ontogénicamente, el páncreas se desarrolla a partir de células progenitoras endodérmicas pluripotentes/ multipotentes que por efecto de factores locales, y de otra naturaleza originan células acinares (exocrinas), ductales (conductos menores que se reúnen en el conducto pancreático o de Wirsung) y células endocrinas. Su origen en el ser humano se inicia en la semana 5-6, observándose ya en la semana 11 los islotes bien delimitados33. La producción de insulina es neta en la semana 20 del embarazo33. En esta época ya parece que se definen cuatro tipos de células endocrinas, células-α;, productoras de glucagón; células-β, productoras de insulina; células-δ, de somatostatina; y por último células PP, productoras de polipéptido pancreático. Al final de la gestación, la mayor cantidad de células son células-β y la menor de células PP (< 2%). Esto se corresponde en líneas generales con las características del páncreas adulto donde el 7080% del islote insular son células-β, el 15-20% células-α, el 5% células-δ y < 2% células PP. Puede por tanto concluirse que el páncreas es un órgano activo al final del primer trimestre y con un papel "decisivo" ya en el cuarto mes del embarazo.
Comentaremos a continuación algunos aspectos que recoge la excelente revisión de Álvarez Escolá y Escrivá Pons99. El tejido endocrino deriva de las células ductales del epitelio pancreático primigenio y se forma mediante neogénesis. Después tales células van formando racimos adheridos al epitelio ductal y la vascularización invade los territorios que están inmaduros, que coexpresan diferentes hormonas pancreáticas y neuropéptidos convirtiéndose posteriormente en islotes de Langerhans100. En los humanos, se definen dos partes, una caudal y otra cefálica, que en consonancia con el movimiento del duodeno el embrión gira y se fusiona con la parte caudal33,101.
En la diferenciación de células primordiales a células endocrinas pancreáticas participan diferentes factores de transcripción, entre los que destaca el factor PDX-1102. También participan otros factores como el PAX, Nk2.2, NK6.1 que se acoplan con el crecimiento celular y la progresiva inervación y vascularización del páncreas55,99. A la formación de las diferentes células endocrinas y expansión de los islotes contribuyen también diferentes factores de crecimiento como PDGF, VEGF, FGF-7, pero particularmente los factores de crecimiento similares a la insulina (IGFs) que se expresan en el estroma pancreático adyacente al epitelio ductal. Así, la especialización a células-β es "canalizada" fundamentalmente por el factor de transcripción PDX-1102. Este PDX-1 interviene también en la maduración de las células-β y en el control de la expresión del gen de la insulina. El factor de transcripción PDX-1 depende a su vez de otros factores de transcripción como el HNF-3β y β-2/Neuro D103.
Como hemos señalado los IGFs, particularmente el IGF-1, son fundamentales para la especialización, crecimiento, maduración de los islotes y por tanto para la producción de insulina. La masa de células-β depende de un equilibrio entre división-crecimiento y muerte por apoptosis. Dicho equilibrio condiciona a su vez la producción de insulina y por tanto la adaptación de las células-β a distintas condiciones ambientales (p.e. carestía, adecuación o exceso de nutrientes). La formación/no formación de células (hiperplasia/aplasia) es un aspecto típico del embarazo, mientras que en el adulto los mecanismos adaptativos atañen fundamentalmente al tamaño celular (hipertrofia/atrofia). Esto sugiere la existencia de plasticidad pancreática que permite responder a situaciones donde la demanda de insulina se incremente99. Recientemente se ha publicado un estudio donde se señala que durante el embarazo en el feto el área de células-β se incrementa, no existiendo cambios en el tamaño de dichas células, ya que es frecuente el incremento del número de islotes pequeños pero no el del número de células-β por islotes. Tampoco se producen cambios en la replicación, pero el número de células ductales positivas para insulina se incrementan durante el embarazo104.
En los roedores, al final de la gestación se incrementa significativamente la masa celular105, posteriormente durante el periodo postnatal tiene lugar un incremento de la apoptosis manteniéndose la neogénesis. En los roedores adultos, la formación de nuevas células-β se reduce de forma marcada, con lo que puede decirse que la masa de células insulares del adulto se ha "prefijado" durante la preñez y la etapa perinatal106.
El ambiente uterino. Clave para un buen desarrollo del páncreas endocrino
Como hemos señalado, el páncreas fetal ya es un órgano activo al final del primer trimestre, momento en el que la placenta es una estructura en evolución hacia la placenta madura. Por tanto los factores histiotróficos que condicionan el crecimiento/desarrollo del embrión van a ser clave para el crecimiento/maduración de la placenta y del páncreas embrionario y fetal. Puede afirmarse que un ambiente anómalo alterará la disponibilidad de sustratos, la angiogénesis y la producción de factores hormonales maternos34 que a su vez redundará en la producción adecuada de factores de transcripción, factores endocrinos y de variada índole, claves para el crecimiento y maduración normal del nuevo ser (fig. 3).
La reducción de la circulación materno-fetal, de la disponibilidad de nutrientes por insuficiencia histiotrófica o placentaria o por alteraciones en la cantidad y calidad de la dieta, la diabetes materna y DG alteran no sólo el crecimiento del ser en formación, sino su tamaño y la capacidad de las células endocrinas pancreáticas, promoviendo a largo plazo ciertas "incapacidades" que predisponen entre otros aspectos a enfermedades degenerativas como la TD2M o el síndrome metabólico6,7,27,32.
El concepto de programación fetal (y por tanto de programación de las capacidades pancreáticas) es el proceso resultante de un estímulo (o agresión) en uno o varios momentos críticos de la gestación que desencadena efectos permanentes que afectan al tamaño, estructura, fisiología y metabolismo. Estas respuestas que facilitan la probabilidad de enfermedades diversas en edades tardías del ser humano, son en definitiva consecuencia de adaptaciones fetales y/o embrionarias que se producen, con gran frecuencia ligadas a desnutrición durante la gestación y que permitirá la supervivencia "económica" del nuevo ser con la puesta en marcha de mecanismos o procesos "ahorradores".
¿Genotipo o fenotipo ahorrador?
Aunque ambos aspectos son concordantes y puedan parecer claramente idénticos, caben algunos matices dignos de ser comentados. El ser humano, como la mayoría de los seres heterótrofos, debe obtener sustratos energéticos, estructurales y reguladores de otros seres en donde se encuentran transformados. No obstante, la oferta de alimentos es en muchos casos no fija y en la mayoría de las situaciones escasa. La evolución de las especies según Faustino Cordón107 parece estar canalizada y dirigida por los medios nutritivos a su alcance; así, un medio estable induce normalmente pequeños cambios adaptativos, mientras que un medio escaso normalmente promueve búsqueda de nuevos nichos ecológicos y de adaptaciones profundas que han llevado a la aparición, mutación, activación o represión de ciertos genes que favorezcan o permitan la existencia y la perpetuidad de la especie.
Genotipo ahorrador. Hace cinco décadas surgió la idea o hipótesis del "Genotipo ahorrador" acuñada por Neel108, según la cual la selección evolutiva favorece a los seres dotados de genes que les permitan acumular reservas metabólicas energéticas en épocas de abundancia de alimentos, para poder afrontar con ventaja las épocas de escasez. Estos genes ahorradores conferirían una predisposición a la acumulación de energía, fundamentalmente en forma de grasa, ya que es un sustrato que rinde más energía por gramo que los otros macronutrientes y que ocupa poco espacio, al almacenarse en forma no hidratada a diferencia del glucógeno o las proteínas62. No debemos olvidar que metabólicamente la grasa además genera más agua por gramo que los hidratos de carbono, o las proteínas, aspecto fundamental en un mundo donde la disponibilidad de agua es a veces muy limitada109-111. También la capacidad metabólica y el rendimiento de la conversión de grasa alimentaria en grasa corporal es muy elevada62. Además, la grasa de los alimentos es tremendamente palatable, es decir los seres dotados de capacidad para detectar alimentos grasos estarían en ventaja respecto a aquellos que no la tuvieran62. Este es un campo muy novedoso y poco conocido, así, se sugiere la existencia de capacidad receptora y de genes implicados en la sensibilidad para el amargo o el dulce, aspectos claves de algunos alimentos de la dieta mediterránea112.
Entre los genes ahorradores se encuentran aquellos que retrasan la utilización de algunos nutrientes (p.e. grasas). Estos genes podrían estar implicados en mecanismos para detectar, absorber, sintetizar, almacenar o retrasar la utilización e hidrólisis de los triglicéridos. Además controlarían la génesis de compuestos controlados por el tejido adiposo y por los núcleos hipotalámicos y de otras regiones del sistema nervioso y de la economía que intervienen a su vez el comportamiento alimentario (p.e. interleuquinas, adipocitoquinas, orexinas y antiorexinas, etc). También serían genes ahorradores los que en presencia reducida de sustratos limitaran la expresión de receptores/transportadores de glucosa, insulina, hormona de crecimiento, IGF, etc., siendo este aspecto de crucial importancia en la propia célula-β. Este tema ha sido mucho menos estudiado, y la información disponible, con mucho, se refiere a mecanismos relacionados con la obesidad y la diabetes, particularmente del adulto. No obstante en la actualidad vamos disponiendo de información creciente al respecto. Aquellos niños SGA, dotados de ciertos polimorfismos en esos genes, tendrían mayor probabilidad de sobrevivir en un mundo donde la disponibilidad de sustratos es escasa o muy cambiante. Entre los genes más estudiados en el adulto se encuentran los genes de la perilipina (PPLIN), del receptor de la leptina (LERP), de la proteína ligadora de ácidos grasos (FABP2), receptor activador y2 de la proliferación de los peroxisomas (PPARy2), de la termogenina (UCP), del sustrato 1 del receptor de la insulina (IRS-1), etc.
Los factores genéticos parecen ser responsables de una gran parte de la varianza asociada con la sensibilidad a la insulina, y por tanto juegan un papel muy importante en el desarrollo de la resistencia a la insulina. Por ello la resistencia a dicha hormona puede ser consecuencia de heredar varias mutaciones en una variedad de genes113. Dado que la T2DM resulta de la interacción de la predisposición genética y se acelera por factores ambientales, puede especularse que diferentes genes con diferente penetrancia pueden contribuir a determinar una alteración prevalente en la acción de la insulina y/o en la secreción de esta hormona114.
Del Prato et al.114 han construido diferentes modelos, considerando la resistencia a la insulina y defectos en la célula-β, para explicar la T2DM y el síndrome metabólico. Según el modelo 1, el síndrome metabólico se desarrolla debido a interacciones entre la resistencia a la insulina y una hiperinsulinemia compensadora. El modelo 2 considera un ambiente genético propicio para el desarrollo de la resistencia a la insulina y defectos en la célula-β, con lo que debido a la secreción insuficiente de insulina se llega a la hiperglucemia y a la diabetes. En el modelo 3, una predisposición genética común origina la acción disminuida de la insulina y el defecto de la célula-β.
El análisis mutacional de la cascada de señalización de la insulina ha identificado un polimorfismo [972G→A (glicina→arginina)] en el codon 972 del ISR-1, siendo la prevalencia de tal polimorfismo del 9% entre los caucásicos114
A nivel del PPARy existen polimorfismos frecuentes que parecen afectar la sensibilidad a la insulina en la población general115. También se ha descrito que los individuos diabéticos y prediabéticos pueden tener una reducción significativa en la expresión de genes que codifican enzimas del metabolismo oxidativo y de la función mitocondrial116. Así, el gen de la calpaina-10 (CAPN10) parece estar relacionado con la resistencia a la insulina. Recientemente algunas variantes genéticas del gen de la proteína asociada a la masa grasa y obesidad o dioxigenasa dependiente de α-cetoglutarato (fat mass and obesity-associated protein también conocido como alpha-ketoglutarate-dependent dioxygenase gene, FTO) se han asociado con la resistencia a la insulina y al síndrome metabólico117,118.
No obstante, la contribución de estos genes sobre la acción/efectos de la insulina es limitada, ya que se ha probado que la resistencia a la insulina está relacionada con mutaciones silenciosas de muchos genes, y de los que cabe esperar también interacción (sinergia o antagonismo). Recientemente se ha producido un gran interés por el factor de transcripción 7-similar al 2 (transcription factor 7-like 2, TCF7L2)119 ya que se ha encontrado una fuerte asociación de este gen con la T2DM en la mayoría de las poblaciones que ha sido confirmada en un metaanálisis realizado en unas 50.000 personas120,121. El polimorfismo CT/TT predice fuertemente el riesgo futuro de T2DM, ya que el alelo de riesgo T se asoció con una secreción reducida de insulina, efectos tipo incretina, y nivel incrementado en la producción de glucosa hepática. Los homocigotos TT tienen incrementado 5 veces la expresión de TCF7L2 en los islotes humanos. Esta sobreexpresión redujo la secreción de insulina tras la estimulación por glucosa. Por tanto puede sugerirse que el riesgo incrementado de T2DM debido a la variante TCF72 atañe al eje enteroinsular, incrementando la expresión del gen en los islotes, y disminuyendo la secreción de insulina121.
También debemos destacar el papel del PPLIN. La perilipina eleva la estabilidad de las gotas de grasa en los adipocitos blancos122 y bloquea, al menos parcialmente, la transformación de los preadipocitos en adipocitos123. Dicha molécula ralentiza o bloquea la acción de la lipasa sensible a hormonas a través de un mecanismo no perfectamente dilucidado que atañe a la proteina tirosina kinasa124. La expresión del PPLIN es importante en poblaciones que padecen de forma habitual hambruna, habiéndose estudiado la relación entre perilipina, resistencia a la insulina y obesidad en el ratón knockout125.
Estudios realizados por Corella et al.126 en pacientes obesos, sugieren que el polimorfismo del PPLIN a nivel 11482 G > A condiciona la respuesta a dietas hipocalóricas y asegura la supervivencia en situaciones de ingesta disminuida de energía. Mientras que los individuos no mutados (GG) responden después de 3 meses o 1 año de tratamiento, reduciendo el peso corporal un 4,4% o un 7,7% respectivamente, los individuos portadores del alelo A (AG y AA) son hiporrespondedores a la dieta de 1.200 kcal y no reducen significativamente su peso corporal después de dicho tratamiento.
Nuestro equipo ha observado que aquellos neonatos que presentan el polimorfismo S19W para el gen de la Apo A5 (APOA5) crecen menos intraútero. Esta situación es más marcada cuando tanto los niños como sus respectivas madres son portadoras W127. La Apo A5 es una proteína que ayuda a la metabolización de las VLDL activando la enzima lipoprotein lipasa (LPL)128. Se conoce que la placenta posee dicha enzima para permitir la cesión de ácidos grasos de los triglicéridos de las VLDL desde la madre a la sangre fetal129. A su vez, estos neonatos muestran una mayor sensibilidad a la insulina (medida como reducción de la resistencia a la insulina) por el marcador de resistencia a la insulina en la homeostasis de la glucosa (HOMA-IR) que aquellos que no presentan la mutación127 (fig. 4). Es interesante observar que los neonatos mostraron una reducción del crecimiento de tipo asimétrico, ya que peso, el índice de masa corporal (IMC) e índice ponderal (IP) estuvieron disminuidos, pero no la longitud corporal (talla), muy posiblemente por deprivación nutricional aguda, particularmente de ácidos grasos, durante el periodo de altísimo crecimiento, ya que la longitud no se afectó130.

Fenotipo ahorrador. El término "Fenotipo ahorrador" atañe a las características de individuos que son capaces de vivir mediante la reducción de la utilización de sustratos. Este término fue acuñado por Barker hacia los años 70 y su hipótesis6,7 es hoy por hoy una de las más citadas en la bibliografía internacional en relación con la programación de enfermedades metabólicas en el adulto. Para Barker et al.6,7 la deficiencia de nutrientes en la etapa perinatal induce una adaptación metabólica cuya finalidad es proteger al órgano central de la economía, el sistema nervioso, con lo que otros tejidos menos exigentes para la existencia del ser vivo se ajustan para reducir la utilización de sustratos. Esta situación será válida mientras se mantenga la deficiencia de nutrientes, pero se presentarán alteraciones metabólicas y funcionales si posteriormente el individuo queda expuesto a condiciones diferentes a las que existían cuando se instauró la "programación"131. La hipótesis de Barker o hipótesis de la "programación metabólica" implica que los cambios que acontecen en etapas muy precoces de la vida, forzadas por condiciones adversas o de limitación, quedan programadas, con lo que la adaptación metabólica se vuelve permanente. Este aspecto parece adquirir enorme importancia en aquellos individuos que siendo "fenotipo ahorradores", son sometidos a catch up forzado o a sobrealimentación postnatal, ya que la situación de abundancia redundaría en la puesta en marcha de mecanismos que incrementarían la resistencia a la insulina y llevarían a un proceso degenerativo conocido como "Síndrome metabólico".
Según Álvarez Escolá y Escrivá Pons99, los mecanismos que determinan las adaptaciones del metabolismo ocurridos han sido plasmados por Langley-Evans132 y señalan como responsables a una serie de cambios en la función placentaria, en el número y tipo de células presentes en los tejidos fetales en pleno desarrollo o a mecanismos epigenéticos que modifican la expresión génica.
Malnutrición o subnutrición materna
Efectos sobre la cantidad de células-β, la producción de insulina y la sensibilidad a dicha hormona. La malnutrición afecta al tamaño del páncreas en humanos133, si bien son muy escasos los datos del tamaño pancreático en gestación105. La nutrición insuficiente "intraútero", bien de tipo primario -por disminución de la ingesta- o secundaria -debido a disminución de la circulación materno-fetal o a insuficiencia placentaria- limita la masa de células-β en los roedores134. No obstante muchos de los mecanismos implicados no son bien conocidos, ya que éticamente no es posible estudiar dichos procesos a nivel celular o de la biología molecular en el páncreas embrionario y fetal humano; y que por otra parte, los modelos animales no siempre mimetizan lo que sucede en humanos. De hecho muchos aspectos de la maduración (p.e. nerviosa) ocurren en los humanos durante la fase tardía de la gestación mientras que tiene lugar en otras especies en la época perinatal o durante la lactancia135. No obstante los modelos animales han sido de gran utilidad para conocer algunas bases fundamentales que relacionan la nutrición materna con el funcionamiento de las células-β pancreáticas.
Así en la rata, la restricción de la ingesta durante la gestación produce perturbaciones metabólicas que modifican el desarrollo de las células fetales productoras de insulina136-138. Dichos islotes son más pequeños, tiene menos células-β que aquellos cuyas madres se alimentaron ad libitum. Tras el nacimiento, en la mayoría de las especies acontece una ola de apoptosis y las células que sufren muerte programada son sustituidas por otras generadas mediante neogénesis, preparando metabólicamente al recién nacido para el periodo postnatal. Sin embargo, en el caso de la DG139 o de madres con diabetes tratadas de forma inadecuada durante el embarazo140 se induce un incremento de la masa de células-β y del contenido pancreático de insulina debido a la instauración de resistencia a la insulina.
Un mal desarrollo del páncreas endocrino debido a malnutrición intraútero puede originar intolerancia a la glucosa y diabetes en la edad adulta, como se ha sugerido en múltiples estudios epidemiológicos141,142, ya que la función de las células de los islotes es liberar cantidades adecuadas de hormonas en respuesta a nutrientes, hormonas y estímulos nerviosos con el objeto de mantener el sustrato fundamental para el sistema nervioso, la glucosa, dentro de límites estrechos. Debemos señalar que la especialización del crecimiento y división de otras células endocrinas insulares son aspectos hoy por hoy muy poco conocidos. No obstante, sabemos que la malnutrición induce elevación de la producción de glucagón en la madre y en el feto, mientras que la de insulina decae. No tenemos referencias sobre cómo se afecta la tasa de células productoras de somatostatina o/y las PP. Dado que el crecimiento celular está regulado también por factores locales como el IGF, la notable caída de la producción de IGF debe afectar la producción de somatostatina en el páncreas.
Como es sabido el regulador principal y directo de la tasa de insulina es la glucosa, teniendo los ácidos grasos, los aminoácidos y los neurotransmisores un papel mucho menor, y casi nulo en ausencia de glucosa. Dicho hidrato de carbono, no sólo regula la secreción sino también su síntesis, requiriéndose la presencia de glucosa en el interior de la célula insular143. Por ello en la actualidad es de gran transcendencia e importancia conocer aspectos específicos de la funcionalidad de los transportadores de glucosa GLUT-1y GLUT-4 y también GLUT-2 y GLUT-3 de la célula pancreática34,36,37,99.
En el periodo fetal, la nutrición inadecuada afecta muy negativamente, tanto en el hombre como en los animales de experimentación, a la funcionalidad de la célula productora de insulina144. Algunos mecanismos propuestos son el daño del desarrollo del páncreas fetal y la incapacidad de respuesta de los islotes fetales aislados138, la disminución de la capacidad de la biosíntesis de la hormona145 y de la secreción de la misma146. La célula-β es muy susceptible a cambios en la disponibilidad de sustratos y hormonas durante el periodo fetal. Dicha célula interacciona con otras células modulando y siendo modulada mediante la expresión de factores de transcripción. Así, la hipoglucemia afecta la expresión de PDX-1147. La hipoglucemia y los ácidos grasos en exceso, entre otros, inhiben la expresión en las células-β de PDX-1 y HNF-3β y en menor medida de β-2148,149.
Los niveles de IGF y de la hormona de crecimiento (GH) regulan el crecimiento insular140. Es por tanto un círculo vicioso, menos IGF, menos células, menos IGF. Es decir la producción de IGF es muy sensible al estado nutricional150.
La subnutrición disminuye la expresión de factores transcripción, lo cual redunda en la disminución del crecimiento fetal y por tanto de la masa de células-β (fig. 5). Esta situación hace al ser vivo que se encuentre no capacitado para responder a situaciones no programadas, ya que como hemos comentado, el número de células-β pancreáticas se mantiene relativamente constante después del nacimiento. Además cuando existe malnutrición con aporte reducido de proteínas, tiene lugar un incremento de la ola apoptótica con la consiguiente disminución de la masa celular-β y de los niveles de IGF151.

No debemos olvidar que la situación se complica, cuando se elevan las concentraciones plasmáticas y tisulares de glucocorticoides. En malnutrición existe normalmente un incremento marcado de los niveles de glucocorticoides (fig. 5). Si bien los glucocorticoides son esenciales para el desarrollo normal del feto, la elevación marcada afecta negativamente al crecimiento, ya que reduce la expresión del factor de transcripción central PDX-1 y reduce la concentración de receptores para IGF-1152.
Sin embargo, la reducción de la capacidad de producir y secretar insulina debida a la reducción de la masa de células-β es compensada, al menos parcialmente, por el incremento de la sensibilidad a la insulina (fig. 5). Este aspecto ha sido señalado en diferentes especies como el mono Rhesus y la rata. Así dietas hipoproteicas y/o hipocalóricas inducen incremento de la sensibilidad a la insulina153,154. Por su parte, es conocido en humanos que durante la malnutrición marásmica, aunque se observa reducción de la producción de insulina y elevación de los glucocorticoides, hay un incremento de la sensibilidad a la insulina155.
Esto nos lleva a completar un poco más el "mosaico" del fenotipo ahorrador poniendo nuevas "teselas", ya que dado que la insulina promueve efectos anabólicos, la hipersensibilidad a la insulina tendría efectos ventajosos en ambientes de escasez. Mientras el estado de subnutrición persiste, los tejidos periféricos responden más intensamente a la insulina, compensándose el déficit hormonal producido por el daño pancreático y se facilita el anabolismo. Esta situación se convertiría en negativa cuando deja de haber una nutrición limitada o reducida (fig. 5).
Como hemos comentado repetidas veces la información disponible en gestación deriva de modelos animales, existiendo información bastante exigua sobre lo que ocurre en fetos vivos viables. Nuestro grupo ha observado en sangre de cordón umbilical que en neonatos a término y sin distrés fetal, objetivizado por el índice de Apgar a tiempo 1 min y 5 min, que en aquellos neonatos con reducción simétrica del crecimiento (peso, talla, IMC, IP) se observan niveles mucho menores de insulina, pero tanto el HOMA-IR como el QUICKI (marcadores de resistencia a la insulina y sensibilidad a la insulina, respectivamente) están respectivamente disminuidos y aumentados (Gesteiro et al., datos sin publicar), apoyando lo comentado en párrafos anteriores.
La alimentación de la madre. Cambios en la sensibilidad a la insulina sin afectar el tamaño del feto
Surgen, después de conocer lo anteriormente expuesto, diferentes cuestiones entre las que destaca ¿Puede la alimentación incorrecta durante la gestación, sin afectar de forma neta al crecimiento fetal, afectar al páncreas endocrino fetal y a la sensibilidad a la insulina? ¿Suelen mantenerse los hábitos dietéticos incorrectos durante todo el crecimiento?
Las características de la dieta, en particular, el tipo de hidratos de carbono156 y su carga glucémica157, la contribución de los AGS al total energético158, la relación AGS/hidratos de carbono88,159, el tipo y cantidad de fibra160, el aporte de ácidos grasos omega-3161 y la relación omega-6/omega-3161 afectan la sensibilidad o resistencia a la insulina de sus consumidores.
Smith et al.159 han mostrado que algunos polimorfismos relacionados con la capacidad del adipocito para almacenar grasa están relacionados con el marcador HOMA-IR de la resistencia a la insulina. Así, estudiaron en una población de mujeres emigrantes de origen indio, malayo y chino en Singapur la respuesta insulinémica a la dieta y valoraron entre otros aspectos la resistencia a la insulina mediante el HOMA-IR. Estos autores encontraron que aquellas mujeres que eran homocigotas para el PPLIN en posición 11482G > A (AA) mostraban incrementos marcados de la resistencia a la insulina a medida que se incrementaba la relación AGS/hidratos de carbono en sus dietas. Este aspecto se demostró de forma separada en los tres grupos étnicos, sugiriendo que aquellos individuos que proceden de regiones donde durante generaciones se ha producido hambruna están muy bien adaptados a dietas ricas en hidratos de carbono. En aquellos individuos cuyas dietas eran más ricas en AGS y más pobres en hidratos de carbono, tenía lugar un incremento de la resistencia a la insulina.
La transferencia de lípidos a través de la placenta cambia según los lípidos presentes en el útero y en la circulación uterina40,41,162,163. Nuevamente existe una información abundante en modelos animales, pero por razones éticas es indirecta y mucho menos abundante en humanos. Así, un incremento de los ácidos grasos a nivel uterino reduce la sensibilidad a la insulina164. Si bien los mecanismos no son bien conocidos, se tiene información de la permanencia de los efectos, ya que una elevación de la concentración uterina de AGS en ratas, eleva de forma permanente la relación glucosa/ insulina en los descendientes165-167, sugiriendo una deficiente utilización de la glucosa. Más recientemente se ha mostrado que la elevación de los AGP omega-3 se relaciona con incremento de la sensibilidad a la insulina, mientras que un exceso de AGP omega-6 perinatal se relaciona con elevación de la prevalencia de obesidad en roedores adultos168. Simopoulous169 ha reportado que existe relación entre la composición en AGP con 20-22 átomos de carbono de los fosfolípidos del músculo esquelético, la sensibilidad/resistencia a la insulina y la obesidad.
Por último la relación omega-6/omega-3 presenta un papel esencial en todo este engranaje. Así en un estudio donde se compararon los efectos de relaciones omega-6/omega-3 muy diferentes (59:1 vs 2:1), las crías cuyas madres recibieron dietas con un cociente elevado pesaban un 50% más al destete, y tenían más grasa y el incremento del tamaño de los adipocitos, persistía durante el periodo adulto168. No sólo eso, se favorecía la transformación de los preadipocitos en adipocitos, aspecto central en el desarrollo e instauración de la obesidad168.
Muy recientemente nuestro grupo88 ha estudiado en una muestra de 35 mujeres en las que se estudiaron las características de la dieta durante el primer trimestre del embarazo y de las que se tenían datos sobre hábitos nutricionales durante toda la gestación, los efectos de la calidad de la dieta sobre marcadores de sensibilidad/resistencia a la insulina en los recién nacidos. La dieta durante el primer trimestre de la gestación se evaluó mediante una encuesta de frecuencia de consumo de 169 items clasificados de acuerdo a grupos de alimentos basada en el estudio enKid170. Los tamaños de raciones se evaluaron mediante la ayuda de fotografías171. La calidad de la dieta de las gestantes se cuantificó utilizando el programa informático DIAL55 que además del contenido en nutrientes y en densidad de nutrientes aportó datos sobre el IAS. A su vez, utilizando el cuestionario de ADM del estudio PREDI-MED86,87, se valoró también la calidad de la dieta durante el primer trimestre. Los resultados no dejan indiferente: Menos de la mitad de las madres consumieron dietas con un IAS mayor de 70, lejos del "score" considerado por el programa DIAL55 de excelente. De forma equivalente sólo un tercio de las madres presentaron una ADM > 7 y ninguna dio valores de 11 o más de ADM. Esto sugiere que la calidad de la dieta de las madres del Estudio Mérida era en términos generales bastante inadecuada. Un análisis detallado de la dieta señaló que el porcentaje de kcal aportado por los hidratos de carbono, la grasa total, los AGS era más adecuado en la dieta de las madres con un IAS > 70 o con ADM > 7. De forma equivalente la relación omega-6/omega 3 y el contenido de fibra fue significativamente mayor en las dietas de madres con mayor ADM. Por último el cociente AGS/Hidratos de carbono apareció del orden de un 30-40% más elevado en las dietas de baja calidad por el IAS y por la ADM (fig. 6).

Cuando se analizaron los efectos de tales dietas sobre las características de los recién nacidos, no se encontraron efectos sobre el peso corporal al nacimiento; sin embargo la insulinemia, el HOMA-IR y la glucemia fueron significativamente mayores en aquellos recién nacidos cuyas madres consumieron dietas de menor calidad valorada tanto por el IAS como por la ADM. Aun más significativo fueron los Odds ratio encontrados que señalan que la probabilidad de presentar un aumento de la resistencia a la insulina o hiperinsulinemia al nacimiento es mucho mayor cuando sus madres consumen dietas con IAS < 70 o con dietas con ADM < 7 que cuando las dietas son más apropiadas (IAS > 70; ADM > 7).
Nuestro trabajo también sugiere la importancia de unos hábitos nutricionales correctos desde antes de la gestación, ya que como hemos comentado, durante los primeros meses de la gestación la alimentación del embrión y feto es histiotrófica e histiotrófica-placentaria, con lo que una alimentación incorrecta ya "programa" cambios metabólicos sutiles relacionados con la resistencia/sensibilidad a la insulina.
Según decía el profesor Grande Covián es más difícil cambiar de hábitos nutricionales que de religión, con lo que si se tienen hábitos nutricionales pregestacionales incorrectos, es muy probable que éstos se mantengan durante todo el embarazo, como ya ha sido corroborado en algun trabajo173. También los datos de nuestro equipo88 así lo señalan, ya que de los trece puntos evaluados en la ADM, podemos señalar que, en términos generales, la dieta sigue manteniendo la misma estructura para la mayoría los 13 puntos, es decir sin cambios. Únicamente en los apartados para el consumo de frutas, legumbres, pescado y dulces y pasteles de origen industrial pueden señalarse algunos cambios, no siempre positivos (p.e. reducción de lácteos), aunque la gran mayoría de las madres no mejoró un punto la evaluación de la calidad de su dieta según la ADM.
Conclusión y futuros estudios
En conclusión, la dieta materna condiciona el crecimiento, maduración y desarrollo fetal y por ende del páncreas. Cuando existe malnutrición/hiponutrición marcada se induce reducción asimétrica o simétrica del feto dependiendo de su duración, con efectos importantes en el tamaño y actividad de páncreas. Dichos cambios son modulables por la existencia de polimorfismos que hacen al futuro ser hipo o hiperrespondedor. Las adaptaciones metabólicas a ingestas restringidas conllevan adaptaciones que incrementan la sensibilidad a la insulina y hacen a los individuos más eficaces en un medio limitado. Esta situación se convierte en "peligrosa", debida a su programación permanente, cuando hay superabundancia. Nuevos estudios señalan que en situaciones de alimentación adecuada respecto al consumo de energía, pero inadecuada bajo el punto de vista de calidad y contribución de nutrientes, se puede afectar negativamente a la resistencia a la insulina del recién nacido, induciendo un perfil prediabético ya desde el nacimiento y muy probablemente en la etapa fetal.
Deben realizarse más estudios para conocer los mecanismos y las etapas más cruciales de esta relación, ya que en la actualidad la dieta española es a todas luces pobre en hidratos de carbono y rica en AGS, habiéndose elevado de forma marcada la relación omega-6/omega-3. A su vez un sondeo de las madres y sus hijos hipo/hiperrespondedores, considerando las características dietéticas previas parece imprescindible. Por ello polimorfismos relacionados con los receptores de insulina, el transporte de ácidos grasos, los transportadores de glucosa, la producción de factores de transcripción, y la producción y secreción de hormonas pancreáticas son fundamentales para entender los mecanismos que relacionan el estado embrionario y fetal con la salud en etapas posteriores de la vida.

Agradecimientos
A las madres y recién nacidos participantes en el Estudio Mérida. Agradecemos a los Servicios de Obstetricia y Ginecología del Hospital Central de Mérida, Badajoz, España su ayuda y asesoramiento.
Referencias
1. Bueno-Lozano G, Sarriá A, Bueno M. Aproximación al diagnóstico de la talla baja. In: Bueno M, ed. Crecimiento y desarrollo humanos y sus trastornos, 2nd edition. Ergón; pp. 122-133, Madrid, 1996. [ Links ]
2. Hernández Rodríguez M, Argente Olivier J. Regulación del crecimiento, la diferenciación y el desarrollo. En: Gil A, ed. Tratado de Nutrición. Tomo III. Nutrición humana en el estado de salud. Martínez de Victoria E, Maldonado J, coordinadores, 2.a edición. Editorial Médica Panamericana; pp. 151-177, Madrid, 2010. [ Links ]
3. Bueno M. Crecimiento y desarrollo humanos. In: Bueno M, ed. Crecimiento y desarrollo humanos y sus trastornos, 2nd edition. Ergón; pp. 3-28, Madrid, 1996. [ Links ]
4. Miras Portugal MT. Presentación. In: Pascual-Leone AM, Medina JM, eds. Desarrollo perinatal: Origen de patologías adultas, Monografía XXIII. Instituto de España. Real Academia Nacional de Farmacia; pp. 15-17; Madrid, 2008. [ Links ]
5. Pascual-Leone AM, Medina JM (eds). Instituto de España. Real Academia Nacional de Farmacia, Monografía XXIII. Madrid, 2008. [ Links ]
6. Barker DJ. Fetal origins of coronary heart disease. BMJ 1995; 311: 171-174. [ Links ]
7. Barker DJ. Early growth and cardiovascular disease. Arch Dis Child 1999; 80:305-307. [ Links ]
8. Palinski W, Napoli C. The fetal origins of atherosclerosis: maternal hypercholesterolemia, and cholesterol-lowering or antioxidant treatment during pregnancy influence in utero programming and postnatal susceptibility to atherogenesis. FASEB J 2002; 16:1348-1360. [ Links ]
9. Napoli C, Glass CK, Witztum JL, Deutsch R, D'Armiento FP, Palinski W. Influence of maternal hypercholesterolaemia during pregnancy on progression of early atherosclerotic lesions in childhood: fate of Early Lesions in Children (FELIC) study. Lancet 1999; 354: 1234-1241. [ Links ]
10. David G, Haegel P. Embriología. En: Tuchman-Duplesis, E. ed. Cuadernos prácticos. Vol I, Toray Masson SA, Barcelona, 1970. [ Links ]
11. Moore KL, Persaud TVN. The developing human: Clinically oriented embryology. Moore KL ed. 7a ed., Sauders W.B, Co. Ltd. Philadelphia, Pennsylvania. 2003. [ Links ]
12. OMS/WHO. Definitions and recommendations. International statistical classifications on diseases. 9th revision. Geneva, 1979; 1: 763-768. [ Links ]
13. Bastida Codina S. Estudio "Toledo": Valores de referencia y factores de riesgo lipoproteico en neonatos. Facultad de Farmacia. Universidad Complutense de Madrid. Tesis doctoral. Madrid, 1992. [ Links ]
14. Espárrago Rodilla M. Estudio "La Serena": Características antropométricas y lipoproteicas de neonatos extremeños. Facultad de Farmacia. Universidad Complutense de Madrid. Tesis doctoral. Madrid, 1997. [ Links ]
15. Thompson JL, Manore MM, Vaughan LA. La nutrición en el ciclo vital: embarazo primer año de vida. En: Thompson JL, Manore MM, Vaughan LA, eds. Nutrición. Pearson Education SA. San Francisco, pp.716, 2008. [ Links ]
16. Barker DJP, Bergmnann RL, Ogra PL, eds. The window of opportunity: pre-pregnancy to 24 months of age. Karger, Nestle Nutrition Institute, pp 1-266, Basel, Switzerland, 2008. [ Links ]
17. Herrera E. Aspectos básicos de las adaptaciones metabólicas en la madre durante la gestación y relaciones materno-fetales. En: Herrera E, ed. Bioquímica perinatal. Fundación Areces, Ceura; pp. 17-39, Madrid, 1988. [ Links ]
18. Herrera E, Ramos Álvarez MP. Papel del tejido adiposo, sensibilidad insulínica e ingesta lipídica en la gestación y su implicación en el riesgo de padecer diabetes en la edad adulta. In: Pascual-Leone AM, Medina JM, eds. Desarrollo perinatal: Origen de patologías adultas. Monografía XXIII. Instituto de España. Real Academia Nacional de Farmacia; pp. 205-238, Madrid, 2008. [ Links ]
19. Institute of Medicine, Food and Nutrition Board. Nutrition during Pregnancy, Parts I and II. Washington, DC. National Academic Press, 1990. [ Links ]
20. Mataix JM, Aranda Ramírez P. Gestación en Nutrición y alimentación humana. En: Mataix Verdú J, ed. II. Situaciones fisiológicas y patológicas. Ergón; pp. 1061-1084, Majadahonda, Madrid, 2009. [ Links ]
21. Florido Navío J, Beltrán Montalván E, Campoy Folgoso, C. Nutrición durante la gestación y la lactancia. En: Gil A, ed. Tratado de Nutrición. Tomo III. Nutrición humana en el estado de salud. Martínez de Victoria E, Maldonado J, coordinadores, 2.a edición. Editorial Médica Panamericana; pp. 133-149, Madrid, 2010. [ Links ]
22. McGanity W, Dawson EB, Van Hook J. Maternal nutrition. En: Shils ME, Olson JA, Shike M, Ross AC, eds. Modern Nutrition in health and disease. 9th ed. Lippincott Williams and Wilkins; pp 811-838, Phyladelphia, 1998. [ Links ]
23. Metzger BE, Coustan DR. Proceedings of the Fourth International Workshop-Conference on Gestational Diabetes. Diabetes Care 1998; 21 (Suppl. 2): B1-B167. [ Links ]
24. Aerts L, Van Assche FA. Animal evidence for the transgenerational development of diabetes mellitus. Int J Biochem Cell Biol 2006; 38: 894-903. [ Links ]
25. Holemans K, Aerts L, Van Assche FA. Fetal growth restriction and consequences for the offspring in animal models. J Soc GynecolInvestig 2003; 10: 292-399. [ Links ]
26. Aerts L, Holemans K, Van Assche FA. Maternal diabetes during pregnancy: consequences for the offspring. Diabetes Metab Rev 1990; 6: 147-167. [ Links ]
27. Serranos-Rios M, Reviriego J, Gutiérrez-Fuentes JA. Classification of Diabetes mellitus: Criteria for diagnosis. En: Serrano-Rios M, Gutiérrez Fuentes JA, eds. Type 2 Diabetes Mellitus. Elsevier; pp.1-23, Barcelona, 2010. [ Links ]
28. American Diabetes Association. Gestational diabetes mellitus (Position statement). Diabetes Care 2004; 27 (Suppl. 1): S88-S90. [ Links ]
29. Kervran A, Guillaume M, Jost A. The endocrine pancreas of the fetus from diabetic pregnant rat. Diabetologia 1978; 15: 387-383. [ Links ]
30. O'Sullivan BA, Henderson ST, Davis JM. Gestational diabetes. J Am Pharm Assoc (Wash) 1998; 38: 364-371; quiz 372-373. [ Links ]
31. Gesteiro E, Bastida S, Sánchez-Muniz FJ. Effects of maternal glucose tolerance, pregnancy diet quality and neonatal insulinemia upon insulin resistance/sensitivity biomarkers in normoweight neonates. Nutr Hosp 2011; 26: 1447-1455. [ Links ]
32. Serranos Rios M, Gutiérrez Fuentes JA. Obesity and Type 2 Diabetes Mellitus. The reciprocal impact. En: Serranos Rios M, Ordovás JM, Gutierrez Fuentes JA, eds. Obesity. Elsevier; pp. 215-232, Barcelona, 2011. [ Links ]
33. Solére M, Haegel P. Embriología. En: Tuchman-Duplesis E, ed. Cuadernos prácticos Vol II. Toray Masson SA, Barcelona, 1970. [ Links ]
34. Myatt L. Placental adaptive responses and fetal programming. J Physiol 2006; 572: 25-30. [ Links ]
35. Monreale de Escobar G, Obregón MJ. Consecuencias de la deprivación de iodo y hormonas tiroideas. En: Pascual-Leone AM, Medina JM, eds. Desarrollo perinatal: Origen de patologías adultas. Monografía XXIII. Instituto de España. Real Academia Nacional de Farmacia; pp. 107-130, Madrid, 2008. [ Links ]
36. Scholl O. Maternal Nutrition before and during pregnancy. En: Barker DJP, Bergmnann RL, Ogra PL, eds. The window of opportunity: pre-pregnancy to 24 months of age. Karger, Nestle Nutrition Institute; pp. 79-89, Basel, Switzerland, 2008. [ Links ]
37. Zorzano Olarte A. Utilización de glucosa durante la vida fetal y su regulación nutricional. En: Pascual-Leone AM, Medina JM, eds. Desarrollo perinatal: Origen de patologías adultas. Monografía XXIII. Instituto de España. Real Academia Nacional de Farmacia; pp. 267-301, Madrid, 2008. [ Links ]
38. Santalucia T, Camps M, castello A, Munoz P, Nuel A, Testar X, Palacin M, Zorzano A. Developmental regulation of GLT-1 (erythroid/Hep G2) and GLUT-4 (muscle/fat) glucose transporter expression in rat heart, skeletal muscle, and brown adipose tissue. Endocrinology 1992; 130: 837-846. [ Links ]
39. Santalucia T, Boheler KR, Brand NJ, Sahye U, Fandos C, Viñals F, Ferre J, Testar X, Palacin M, Zorzano A. Factors involved in GLUT-1 glucose transporter gene transcription in cardiac muscle. J Biol Chem 1999; 274: 17626-17634. [ Links ]
40. Larqué E, Demmelmair H, Gil-Sánchez A, Prieto-Sánchez MT, Blanco JE, Pagán A, et al. Placental transfer of fatty acids and fetal implications. Am J Clin Nutr 2011; 96 (Suppl.): 1908S-1913S. [ Links ]
41. Gil-Sánchez A, Koletzko B, Larqué E. Current understanding of placental fatty acid transport. Curr Opin Clin Nutr Metab Care 2012; 15: 265-272. [ Links ]
42. Magnusson-Olsson AL, Hamark B, Ericsson A. Gestational and hormonal regulation of human placental lipoprotein lipase. J Lipid Res 2006; 47: 2551-2561. [ Links ]
43. Wagner CL, Taylor SN, Dawodu A, Johnson DD, Hollis BW. Vitamin D and its role during pregnancy in attaining optimal health of mother and fetus. Nutrients 2012; 4: 208-230. [ Links ]
44. Christesen HT, Falkenberg T, Lamont RF, Jorgensen JS. The impact of vitamin D on pregnancy: a systematic review. Acta Obstet Gynecol Scand 2012; 901: 1357-1367. [ Links ]
45. Martínez Augustin O, Sánchez de Medina López-Huertas F, Suárez Ortega MD. Vitamina D. En: Gil A, ed. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 571-592, Madrid. 2010. [ Links ]
46. Ordovás, JM, Corella D. La revolución del genoma humano. ¿Qué significa genómica, epigenética, nutrigenética, nutrigenómica, metabolómica? En: Vaquero, MP, coordinadora. Genética, nutrición y enfermedad. Instituto Tomás Pascual y Consejo Superior de Investigaciones Científicas; pp. 17-29, Madrid, 2008. [ Links ]
47. Sánchez-Muniz FJ, Bastida S. Nutrigenómica y nutrigenética. Desde la expresión génica a las dietas a medida. Unidad 1. Módulo 3. In: Experto en Nutrición y Planificación Dietética. Departamento de Nutrición y Bromatología I (Nutrición). Facultad de Farmacia. Universidad Complutense de Madrid. COINSA, Madrid, 2010. [ Links ]
48. Sánchez-Muniz FJ, Nus M. Importancia de la interacción dieta-genética en la prevención cardiovascular. Capítulo 7. En: Vaquero P, coordinadora. Genética, nutrición y enfermedad. Instituto Tomás Pascual Sanz y CSIC Editores Médicos, S.A.; pp. 125-144; Madrid, 2008. [ Links ]
49. Mataix Verdú J, Sánchez de Medina Contreras F. Bases metabólicas y moleculares de la nutrición. III. Regulación de la expresión génica. En: Mataix Verdú J, ed. Nutrición y alimentación humana. I. Nutrientes y alimentos. Ergón; pp. 63-69, Majadahonda, Madrid. 2009. [ Links ]
50. Gil Hernández A, Aguilera García C, Gómez Llorente C. Nutrigenómica. En: Gil A, ed. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina F, coordinador, 2.a edición. Editorial Médica; pp. 749-806, Panamericana. Madrid. 2010. [ Links ]
51. SEN, Departamento de Nutrición. Ingestas recomendadas de energía y nutrientes para la población Española (revisadas 2008). En: Moreiras O, Carbajal A, Cabrera L, Cuadrado C, eds. Tablas de Composición de Alimentos. Pirámide. Madrid. 2008. [ Links ]
52. Institute of Medicine, Food and Nutrition Board. Dietary reference intakes: The essential guide to nutrient requirements. Washington DC. National Academic Press, 2006. [ Links ]
53. Institute of Medicine, Food and Nutrition Board. Dietary reference intakes research synthesis workshop summary. Washington DC. National Academic Press, 2006. [ Links ]
54. Gil Hernández A, Mañas Almendros M, Martínez de Victoria Muñoz E. Ingestas dietéticas de referencia, objetivos nutricionales y guías. En: Gil A, ed. Tratado de Nutrición. Tomo III. Nutrición humana en el estado de salud. Martínez de Victoria E, Maldonado J, coordinador, 2a edición. Editorial Médica Panamericana; pp. 31-64, Madrid, 2010. [ Links ]
55. Ortega RM, López-Sobaler AM, Andrés P, Requejo AM, Aparicio A, Molinero LM. DIAL software for assessing diets and food calculations. Departamento de Nutrición (UCM) and Alce Ingeniería SA Madrid, 2004. http://www.alceingenieria.net/nutricion.htm [ Links ]
56. FAO/WHO. The Joint FAO/WHO expert consultation on fats and fatty acids in human nutrition. FAO food and nutrition paper 91. Food and Agriculture Organization of the United Nations, Rome, 2010. ISSN 0254-4725. [ Links ]
57. Díaz L, Zambrano B, Chacón G, Rocha A, Diaz S. Toxoplasmosis y embarazo. Rev Obstet Ginecol Venez 2010; 70: 190-205. [ Links ]
58. Herrera E, Testar X, Llovera M. Efectos metabólicos del etanol. Alcohol en gestación y sus consecuencias en las relaciones feta/madre. En: Herrera E, ed. Bioquímica perinatal. Fundación Areces. Ceura; pp. 577-599, Madrid, 1988. [ Links ]
59. López-Tejero D, Llovera M, Herrera E. Consecuencias del alcohol durante la gestación sobre el desarrollo postnatal. En: Herrera E, ed. Bioquímica perinatal. Fundación Areces. Ceura; pp. 601-627, Madrid, 1988. [ Links ]
60. Sánchez de Medina Contreras F. Metabolismo del alcohol y de otros componentes de los alimentos. En: Gil A, editor. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 735-747, Madrid, 2010. [ Links ]
61. Agencia Española de Seguridad Alimentaria y Sanidad (AESAN). Recomendaciones de consumo de algunas especies de pescado azul. Abril, 2011. http://www.europapress.es/salud/noticia [ Links ]
62. Sánchez-Muniz FJ. Lípidos. En: García-Arias MT, García Fernández MC, eds. Nutrición y Dietética. Universidad de León. Secretariado de Publicaciones y Medios Audiovisales; pp. 119-133. León, 2003. [ Links ]
63. Sánchez-Muniz FJ, Mataix Verdú, J. Aceites y alimentos ricos en grasa. Unidad 5. Módulo 1 Alimentos como herramientas en la planificación dietética. En: Experto en Nutrición y Planificación Dietética. Departamento de Nutrición y Bromatología I (Nutrición). Facultad de Farmacia. Universidad Complutense. COINSA, 2007 y actualizaciones posteriores (2009, 2010, 2011). ISBN-13: 978-84-692-6575-8. [ Links ]
64. Larqué E, Gil-Sánchez A, Prieto-Sánchez MT, Koletko B. Omega 3 fatty acids, gestation and pregnacy otcomes. Br J Nutr 2012; 107: S77-S84. [ Links ]
65. Valenzuela Bonanome A, Uauy Dagach R. Funciones y metabolismo de los ácidos graso esenciales y de sus derivados activos. En: Gil A, ed. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 303-320, Madrid, 2010. [ Links ]
66. Christesen HT, Elvander C, Lamont RF, J0rgensen JS. The impact of vitamin D in pregnancy on extraskeletal health in children: A systematic review. Acta Obstet Gynecol Scand 2012; 901: 1368-1380. [ Links ]
67. da Rocha CM, Kac G. High dietary ratio of omega-6 to omega-3 polyunsaturated acids during pregnancy and prevalence of post-partum depression. Matern Child Nutr 2012; 8: 36-48. [ Links ]
68. Ortega Anta R, Mena Valverde MC, Andrés Carvajales P. Vitamina A. En: Gil A, ed. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 547-569, Madrid, 2010. [ Links ]
69. Rivero Urgell M. Alimentación y nutrición durante el embarazo. In: Mataix J, coordinator. Nutrición y Dietética. Aspectos sanitarios. PLENUFAR. Colegio General de Colegios Oficiales Farmacéuticos; pp. 361-379, Madrid. 1993. [ Links ]
70. Olivares Grohnert M, Arredondo Olguín M, Pizarro Aguirre F. Hierro. En: Gil A, ed. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 669-686, Madrid, 2010. [ Links ]
71. World Health Organization. Assessment of iodine deficiency disorders and monitoring their elimination. Department of Nutrition. Geneva, Switzerland, 2001. [ Links ]
72. Barbel P, Obregón MJ, Bernal J, Escobar del Rey F, Monreale de Escobar G. Iodine supplementation during pregancy: a public health challenge. Trends Endocrin Metab 2007; 18: 338-343. [ Links ]
73. Hoyo C, Murtha AP, Schildkraut JM, Forman MR, Calingaert B, Demark-Wahnefried W, et al. Folic acid supplementation before and during pregnancy in the Newborn Epigenetics STudy (NEST). BMC Public Health 2011; 11: 46. [ Links ]
74. Greenberg JA, Bell SJ, Guan Y, Yu Y-h. Folic acid supplementation and pregnancy: more than just neural tube defect prevention. Rev Obstet Gynecol 2011; 4: 52-59. [ Links ]
75. Paz R de, Hernández Navarro F. Manejo, prevención y control de la anemia megaloblástica secundaria a déficit de ácido fólico. Nutr Hosp 2006; 21:113-119. [ Links ]
76. Varela-Moreiras G, Escudero JM, Aloso-Aperte E. Homocisteína, vitaminas relacionadas y estilo de vida en personas de edad avanzada: estudio Séneca. Nutr Hosp 2007; 22: 363-370. [ Links ]
77. Sánchez de Medina Contreras F. Vitaminas con función de coenzimas. En: Gil A, editor. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 501-524, Madrid, 2010. [ Links ]
78. Ramírez Tortosa MC, Quiles Morales JL. Vitamina C, Vitamina E y coenzima Q. En: Gil A, editor. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 481-500, Madrid, 2010. [ Links ]
79. Alonso Aperte E, Gregorio Varela Mosquera. Acido fólico y Vitamina B12. En: Gil A, editor. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 525-546, Madrid, 2010. [ Links ]
80. Sánchez-Muniz FJ. Aceite de oliva, clave de vida en la Cuenca Mediterránea. An R Ac Nac Far 2007; 73: 653-692. [ Links ]
81. Márquez-Sandoval F, Bulló M, Vizmanos B, Casas-Agustench P, Salas-Salvadó J. Un patrón de alimentación saludable: la dieta mediterránea tradicional. Antropo 2008; 16: 11-22. [ Links ]
82. Serra-Majem L, Trichopoulou A, Ngo de la Cruz J, Cervera P, García Álvarez A, La Vecchia C, et al, on behalf of the International Task Force on the Mediterranean Diet. 2004 Foreword: Does the definition of the Mediterranean diet need to be updated? Public Health Nutr 2004; 7: 927-929. [ Links ]
83. Buckland G, Bach-Faig A, Serra-Majem L. Obesity and the Mediterranean diet: a systematic review of observational and intervention studies. Obes Rev 2008; 9: 582-593. [ Links ]
84. Trichopoulou A, Costacou T, Bamia C, Trichopoulos D. Adherence to a Mediterranean diet and survival in a Greek population. N Engl J Med 2003; 348: 2599-2608. [ Links ]
85. Trichopoulou A, Bamia C, Trichopoulos D. Mediterranean diet and survival among patients with coronary heart disease in Greece. Arch Intern Med 2005; 65: 929-935. [ Links ]
86. Estruch R, Martínez-González MA, Corella D, Salas-Salvadó J, Ruiz-Gutiérrez V, Covas MI et al; PREDIMED Study Investigators. Effects of a Mediterranean-style diet on cardiovascular risk factors: a randomized trial. Ann Intern Med 2006; 145: 1-11. [ Links ]
87. Salas-Salvadó J, Bulló M, Babio N, Martínez-González MÁ, Ibarrola-Jurado N, Basora J et al; PREDIMED Study Investigators. Reduction in the incidence of type 2 diabetes with the Mediterranean diet: results of the PREDIMED-Reus nutrition intervention randomized trial. Diabetes Care 2011; 34: 14-19. [ Links ]
88. Gesteiro E, Rodríguez-Bernal B, Bastida S, Sánchez-Muniz FJ. Maternal diets with low healthy eating index or Mediterranean diet adherence scores are associated with high cord-blood insulin levels and insulin resistance markers at birth. Eur J Clin Nutr 2012; 66: 1008-1015. [ Links ]
89. Kennedy ET, Ohls J, Carlson S, Fleming K. The Healthy Eating Index: design and applications. J Am Diet Assoc 1995; 95:1103-1108. [ Links ]
90. Hahn CS, Rock CL, King I, Drewnowski A. Validation of de Healthy Eating Index with use of plasma biomarkers in a clinical sample of women. Am J Clin Nutr 2001; 74: 479-486. [ Links ]
91. Weinstein SJ, Vogt TM, Gerrior SA. Healthy Eating Index scores are associated with blood nutrient concentrations in the Third National Health and Nutrition Examination Survey. J Am Diet Assoc 2004; 104: 576-584. [ Links ]
92. Koning L, Chiuve SE, Fung TT, Willett WC, Rimm EB, Hu FB. Diet-quality scores and the risk of type 2 diabetes in men. Diabetes Care 2011; 34: 1150-1156. [ Links ]
93. McCullough ML, Feskanich D, Stampfer MJ, Giovannucci EL, Rimm EB, Hu FB, et al. Diet quality and major chronic disease risk in men and women: moving toward improved dietary guidance. Am J Clin Nutr 2002; 76: 1261-1271. [ Links ]
94. Bodnar LM, Siega-Riz AM. A Diet Quality Index for pregnancy detects variation in diet and difference by sociodemographic factors. Public Health Nutr 2002; 5: 801-809. [ Links ]
95. Martínez de Victoria Muñoz E, Mañas Almendros M, Yago Torregrosa MD. Fisiología de la digestión. En: Gil A, editor. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 171-201, Madrid, 2010. [ Links ]
96. Mataix Verdú J, Martínez de Victoria ME. Bases fisiológicas digestivas. I. Sistema digestivo. Bases fisiológicas. En: Mataix Verdú J, ed. Nutrición y alimentación humana. I. Nutrientes y alimentos. Ergón; pp. 5-27, Majadahonda, Madrid, 2009. [ Links ]
97. Mataix Verdú J, Sánchez de Medina Contreras F. Bases metabólicas y moleculares de la nutrición. I. Metabolismo y su regulación. In: Mataix Verdú J, ed. Nutrición y alimentación humana. I. Nutrientes y alimentos. Ergón; pp. 45-62, Majadahonda, Madrid. 2009. [ Links ]
98. Martínez Augustin O, Daddaoua A, Suárez Ortega MD. Relaciones metabólicas tisulares en el ciclo de ayuno y realimentación. En: Gil A, editor. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 405-427, Madrid, 2010. [ Links ]
99. Álvarez Escolá C, Escrivá Pons F. Influencia de la subnutrición perinatal sobre el desarrollo de las células beta y la acción de la insulina: relación con la diabetes 2 adulta. En: Pascual-Leone AM, Medina JM, eds. Desarrollo perinatal: Origen de patologías adultas. Monografía XXIII, Instituto de España. Real Academia Nacional de Farmacia; pp. 239-265, Madrid, 2008. [ Links ]
100. Reusens B, Remacle C. Programming of the endocrine pancreas by the early nutritional environment. Int J Biochem Cell Biol 2006; 38: 913-922. [ Links ]
101. Poirier J, Cohen I, Baudet J, eds. Embriología humana. Versión española de Gómez Sánchez J. Editorial Marbán, Madrid. 1974. [ Links ]
102. Sander M, German MS. The beta cell transcription factors and development of the pancreas. J Mol Med 1997; 75: 327-340. [ Links ]
103. Hediger ML, Overpeck MD, McGlynn A, Kuczmarski RJ, Maurer KR, Davis WW. Growth and fatness at three to six years of age in children born with intrauterine growth retardation. J Clin Endocrinol Metab 2000; 85: 1401-1406. [ Links ]
104. Kaung HG. Growth dynamic of pancreatic islet cell populations during fetal and neonatal development of the rat. Dev Dyn 1994; 200: 163-175. [ Links ]
105. Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010; 53: 2167-2176. [ Links ]
106. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429: 41-46. [ Links ]
107. Cordón F. La alimentación, base de la biología evolucionista. Historia natural de la acción y experiencia. Parte Primera. Origen, naturaleza y evolución del protoplasma. Alfaguara, Madrid, 1978. [ Links ]
108. Neel JV. Diabetes mellitus: a "thifty" genotype rendered detrimental by "progress"? Am J Hum Genet 1962; 14: 352-362. [ Links ]
109. Mataix Verdú J, García Torres L. Agua y equilibrio hidroelectrolítico. En: Mataix Verdú J, ed. Nutrición y alimentación humana. II. Situaciones fisiológicas y patológicas. 2nd edition. Ergón; pp 927-950, Majadahonda, Madrid, 2009. [ Links ]
110. Mataix Verdú J, Carrazo Marín E. Agua. En: Mataix Verdú J, ed. Nutrición para educadores. 2nd edition. Díaz de Santos; pp 183-199, Madrid, 2005. [ Links ]
111. López Novoa JM, López Hernández F. Metabolismo hidromineral: agua y electrolitos. En: Gil A, editor. Tratado de Nutrición. Tomo I. Bases fisiológicas y bioquímicas de la nutrición. Sánchez de Medina E, coordinador, 2.a edición. Editorial Médica Panamericana; pp. 593-621, Madrid. 2010. [ Links ]
112. Arellano Campos O. Percepción del sabor, dieta mediterránea y nutrigenética: efecto sobre componentes del síndrome metabólico. Doctorado en Biotecnología. Facultad de Ciencias Biológicas. Universitat de Valencia. Valencia, 2012. [ Links ]
113. Pedersen O. Genetics of insulin resistance. Exp Clin Endocrin Diabetes 1999; 107: 113-118. [ Links ]
114. Del Prato S, Pulizzi N, Lupi R, Penno G, Miccoli R. Type 2 Diabetes: Insulin resistance v. beta-cell defect. En: Serranos Rios M, Gutiérrez Fuentes JA, eds. Type 2 Diabetes Mellitus. Elsevier; pp. 131-149, Barcelona, 2010. [ Links ]
115. Meirhaeghe A, Amoouyel P. Impact of genetic variation of PPARSgamma in humans. Mol Genet Metab 2004; 83: 93-102. [ Links ]
116. Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, et al. Coordinated reduction of genes of oxidation metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF. Proc Natl Acad Sci USA 2003; 100: 8466-8471. [ Links ]
117. Jacobsson JA, Klñovins J, Kapa I, Danielsson P, Svenssson V, Ridderstrale M, et al. Novel genetic variant in FTO influenes insulin levels and insulin resistance in severely obese children and adolescents. Int J Obes (Lond) 2008; 32: 1730-1735. [ Links ]
118. Do R, Bailey SDS, Desbiens K, Belisle A, Montpetit A, Bouchard C et al. Genetic variants of FTO influence adiposity, insulin sensitivity, leptin levels, and resting metabolic rate in the Quebec Family Study. Diabetes 2008; 57: 1147-1150. [ Links ]
119. Cauchi S, El Achhab Y, Choquet H, Din C, Krempler F, Weitgasser R et al. TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. J Mol Med 2007; 85: 777-782. [ Links ]
120. Florez JC. The new type 2 diabetes gene TCF7L2. Curr Opin Clin Nutr Metab Care 2007; 10: 391-396. [ Links ]
121. Lyssenko V, Lupi R, Marchetti P, Del Guerra S, Orho-Melander M, Almgren P et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest 2007; 117: 2155-2163. [ Links ]
122. Londos C, Gruia-Gray J, Brasaemle DL, Rondinone CM, Takeda T, Dwyer NK, et al. Perilipin: possible roles in structure and metabolism of intracellular neutral lipids in adipocytes and steroidogenic cells. Int J Obes Relat Metab Disord 1996; 20 (Suppl. 3): 97-101. [ Links ]
123. Martínez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, et al. Absence of perilipin results in leenness and reverse obesity in Lepr (db/db) mice. Nat Genet 2000; 26: 474-479. [ Links ]
124. Sztalryd C, Xu G, Dorward H, Contreras JA, Kimmel AR, Londos C. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic action. J Cell Biol 2003; 161:1093-1103. [ Links ]
125. Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O et al. PLIN ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci USA 2001; 98: 6494-6499. [ Links ]
126. Corella D, Qi, L, Tai ES, Deurenberg M, Tan CE, Chew SK, Ordovas JM. Perilipin gene variation determines higher susceptibility to insulin resistance in Asian women when consuming a high-saturated fat, low-carbohydrate diet. Diabetes Care 2006; 29: 1313-1319. [ Links ]
127. Gesteiro E, Bastida S, Sánchez-Muniz FJ. Effects of APOAS S19W polymorphism on growth, insulin sensitivity and lipoproteins in normoweight neonates. Eur J Ped 2011; 170: 1551-1558. [ Links ]
128. Charriere S, Bernard S, Aqallal M, Merlin M, Billon S, Perrot L, et al. Association of APOA5-1131T > C and S19W gene polymorphisms with both mild hypertriglyceridemia and hyperchylomicronemia in type 2 diabetic patients. Clin Chim Acta 2008; 394: 99-103. [ Links ]
129. Magnusson AL, Waterman IJ, Wennergren M, Jansson T, Powell TL. Triglyceride hydrolase activities and expression of fatty acid binding proteins in the human placenta in pregnancies complicated by intrauterine growth restriction and diabetes. J Clin Endocrinol Metab 2004; 89: 4607-4614. [ Links ]
130. Pérez-González JM, Ventura MP, Garagorri JM, Bueno-Lozano O. Retraso de crecimiento intrauterino. En: Bueno M, ed. Crecimiento y desarrollo humanos y sus trastornos. 2nd edition. Ergón; pp. 161-172, Madrid, 1996. [ Links ]
131. Hales CN, Barker DJP. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 1992; 35: 595-601. [ Links ]
132. Langley-Evans SC. Developmental programming of health and disease. Proc Nutr Soc 2006; 65: 97-105. [ Links ]
133. El-Hodhod MA, Nassar MF, Hetta OA, Gomaa SM. Pancreatic size in protein energy malnutrition: a predictor of nutritional recovery. Eur J Clin Nutr 2005; 59: 467-473. [ Links ]
134. Winick M, Noble A. Cellular response in rats during malnutrition at various ages. J Nutr 1996; 89: 300-306. [ Links ]
135. Caravaca Rodríguez FP, Castel Genís JM, Guzmán Guerrero JL, Delgado Pertíñez M, Mena Guerrero Y, Alcalde Aldea MJ, González Redondo P. Bases de producción animal. Catálogo de Publicaciones de la Universidad de Sevilla. Serie manuales Universitarios, n.o 61. Sevilla, 2003. [ Links ]
136. Berney DM, Desay M, Palmer DJ, Greewald S, Brown A, Hales CN, Berry CL. The effect of maternal protein deprivation on the fetal rat pancreas: major structural changes and their recuperation. J Pathol 1997; 183: 109-115. [ Links ]
137. Snoeck A, Remacle C, Reusens B, Hoett JJ. Effect of low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol Neonate 1990; 57: 107-118. [ Links ]
138. Dahri S, Snoek A, Reusens-Billen B, Remacle C, Hoett JJ. Islet function in offspring of mothers on low protein diet during gestation. Diabetes 1991; 40: 115-120. [ Links ]
139. Aerts L, Van Assche FA. Rat foetal endocrine pancreas in experimental diabetes. J Endocrinol 1997; 73: 339-346. [ Links ]
140. Hill JD, PetriK J, Arany E. Growth factors and the regulation of fetal growth. Diabetes Care 1998; 21 (Suppl. 2): 60B-69B. [ Links ]
141. Stanner SA, Bulmer K, Andres C, Lantseva OE, Borodina V, Poteen VV, Yudkin JS. Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrade siege study. Br Med J 1997; 315: 1142-1149. [ Links ]
142. Lumey LH, Stein AD, Kahn HS, van der PAL-de Bruin KM, Blauw GJ, Zybert PA, Susser ES. Cohort profile: the Dutch hunger winter family study. Int J Epidemiol 2007; 36: 1196-1204. [ Links ]
143. Hernández Rodríguez M, Argente Oliver J. Regulación del crecimiento, la diferenciación y el desarrollo. En: Gil A, ed. Tratado de Nutrición. Nutrición Humana en el estado de Salud. Tomo III. Martínez de Victoria E, Maldonado J, coordinadores, 2.a edición. Editorial Médica Panamericana; pp. 151-177, Madrid, 2010. [ Links ]
144. Rao RH. Diabetes in the undernourished: coincidence or consequence? Endocr Rev 1988: 9: 67-87. [ Links ]
145. Arantes VC, Teixeira VP, Reis MA, Latorraca MQ, Leite AR, Carneiro EM, et al. Espression of PDX-1 is reduced in pancreatic islets from pups of rat dams fed a low protein diet during gestation and lactation. J Nutr 2002; 132: 3030-3035. [ Links ]
146. Wilson MR, Hughes SJ. Impaired glucose-stimulated insulin release in islets from adult rats malnourished during foetal-neonatal life. Mol Cell Endocrinol 1998: 142: 41-48. [ Links ]
147. Petersen HV, Peshavaria M, Pedersen AA, Philippe J, Stein R, Madsen OD, Serup P. Glucose stimulates the activation domain potential of the PDX-1 homeodomain transcription factor. FEBS Lett 1998; 431: 362-366. [ Links ]
148. MacFarlane WM, Read ML, Gilligan M, Bujalska I, Docherty K. Glucose modulates the binding activity of the beta-cell transcription factor IUF1 in a phosphorylation-dependent manner. Biochem J 1994; 303: 625-631. [ Links ]
149. Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patané G, Laybutt R, et al. Increased expression of antioxidant and antiapoptotic genes in islets that may contribute to beta-cell survival during chronic hyperglycemia. Diabetes 2002; 51: 413-423. [ Links ]
150. Petrick J, Arany E, Mc Donald TJ, Hill DJ. Apoptosis in the pancreatic islet cells of the neonatal rat is associated with a reduced expression of insulin-like growth factor II that may act as a survival factor. Endocrinology 1998; 139: 2994-3004. [ Links ]
151. Petrick J, Reusens B, Arany E, Remacle C, Coelho C, Hoet JJ, Hill DJ. A low protein diet alters the balance of islet cell replication growth factor-II. Endocrinology 1999; 140: 4861-4873. [ Links ]
152. Arumugam R, Horowitz E, Lu D, Collier JJ, Ronnebaum S, Fleenor D, Freemark M. The interplay of prolactin and the glucocorticoids in the regulation of beta-cell gene expression, fatty acid oxidation, and glucose-stimulated insulin secretion: Implications for carbohydrate metabolism in pregnancy. Endocrinology 2008 149: 5401-5414. [ Links ]
153. Kemmitz JW, Roecker EB, Weindruch R, Elson DF, Baum ST, Bergman RN. Dietary restriction increases insulin sensitivity and lowers blood glucose in rhesus monkeys. Am J Physiol 1994; 29: E540-E547. [ Links ]
154. Cartee GD, Kietzke EW, Briggs-Tung C. Adaptation of muscle glucose transport with caloric restriction in adult, middle-aged and olds rats. Am J Physiol 1994; 266: R1443-R1447. [ Links ]
155. Bowie MD. Intraveneous glucose tolerance in kwashiorkor and marasmus. S Afr Med J 1964; 38: 328-329. [ Links ]
156. Burger KNJ, Beulens JWJ, Boer JMA, Spijkerman AMW, van der A.DL. Dietary glycemic load and glycemic index and risk of coronary heart disease and stroke in Dutch men and women: The EPIC-MORGEN Study. PLoS One 2011; 6 (10): e25955. [ Links ]
157. Livesey G. A systematic review of the glycaemic response to foods and health. ILSI Europe workshop. Glycaemic response on health; pp. 82-127, Nice, France. 6-8 Dec 2006. [ Links ]
158. López S, Bermúdez B, Pacheco YM, Villar J, Abia R and Muriana FJG. Distinctive postprandial modulation of beta-cell function and insulin sensitivity by dietary fats: monounsaturated compared with saturated fatty acids. Am J Clin Nutr 2008; 88: 638-644. [ Links ]
159. Smith CE, Arnett DK, Corella D, Tsai MY, Lai CQ, Parnell LD, et al. Perilipin polymorphism interacts with saturated fat and carbohydrates to modulate insulin resistance. Nutr Metab Cardiovasc Dis 2012; 22: 449-455. [ Links ]
160. Sánchez-Muniz FJ. Dietary fiber and cardiovascular health. Nutr Hosp 2012; 27: 40-54. [ Links ]
161. Martín de Santa Olalla L, Sánchez-Muniz FJ, Vaquero MP. N-3 fatty acids in glucose metabolism and insulin sensitivity. Nutr Hosp 2009; 24: 203-217. [ Links ]
162. Herrera E, Lasunción MA. Maternal-fetal transfer of lipid metabolites. En: Polin RA, Fox WW, Abman SH, eds. Fetal and neonatal physiology. Saunders; pp 2855-2865, Philadelphia, 2004. [ Links ]
163. Herrera E, López-Soldado I, Limones M, Amusquivar E, Ramos MP. Lipid metabolism during the perinatal phase and its implication on postnatal development. Int J Nutr Vit Res 2006; 76: 216-224. [ Links ]
164. Amusquivar E, Rupérez FJ, Barbas C, Herrera E. Low arachidonic acid rather than alpha-tocopherol is responsible for the delayed postnatal development in offspring of rats fed fish oil instead olive oil during pregnancy and lactation. J Nutr 2000; 130:2855-2865. [ Links ]
165. Gerber RT, Holemans K, O'Brien-Coker I, Mallet AI, van Bree R, Van Assche FA, Poston L. Cholesterol-independent endothelial dysfunction in virgin and pregnant rats fed a diet high in saturated fat. J Physiol 1999; 517: 607-616. [ Links ]
166. Guo F, Jen KL. High-fat feeding during pregnancy and lactation affects offspring metabolism in rats. Physiol Behav 1995; 57: 681-686. [ Links ]
167. Khan IY, Dekou V, Douglas G, Jensen R, Hanson MA. A high-fat diet during pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am J Physiol Regn Intergr Comp Physiol 2005; 288: R127-R133. [ Links ]
168. Ailhaud G, Guesnet P. Fatty acid composition of fats is an early determinant of childhood obesity; a short review and an opinion. Obes Rev 2004; 5: 21-26. [ Links ]
169. Simopoulo AP. Fatty acid composition of the skeletal muscle membrane phospholipids, insulin resistance and obesity. Nutr Today 1994; 2: 12-16. [ Links ]
170. Serra Majem L, Aranceta Bartrina J. Alimentación infantil y juvenil. Estudio enKid. Ed. Masson. Barcelona, 2002. [ Links ]
171. Mataix Verdú J, Llopis González J. Manual gráfico e contenido nutricional de pratos galegos. Jiménez Contreras JF, Lendoiro Otero RM, Meniño Olivera M, eds. Consellería de Sanidad, Carrefour, Galicia, España, 1993. [ Links ]
172. Cervera Ral P, Fernández-Ballart D. Alimentación, embarazo y lactancia. En: Guías alimentarias para la población española. Recomendaciones para una vida saludable, SENC, IM & C, SA, pp. 355-364, Madrid, 2001. [ Links ]
173. Crozier SR, Robinson SM, Godfrey KM, Cooper C, Inskip HM. Women's dietary patterns change little from before to during pregnancy. J Nutr 2009; 139: 1956-1963. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Francisco J. Sánchez-Muniz.
Departamento de Nutrición y Bromatología I (Nutrición).
Facultad de Farmacia.
Universidad Complutense de Madrid.
28040 Madrid. España.
E-mail: frasan@farm.ucm.es
Recibido: 9-XI-2012.
Aceptado: 12-XII-2012.

