










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
Presentamos el caso de un paciente con antecedentes de polineuropatía amiloidótica familiar por transtiretina (TTR-FAP) remitido a la consulta de nutrición por pérdida ponderal y que, en el proceso de evaluación nutricional, es diagnosticado de hipocupremia severa. Se revisan las causas más importantes del déficit de cobre y su posible relación con la TTR-FAP, así como sus manifestaciones clínicas y tratamiento.
CASO CLÍNICO
Se trata de un varón de 79 años, exfumador, portador de prótesis dental, hipertenso, dislipidémico y diabético de tipo 2 con más de 10 años de evolución, sin complicaciones conocidas y con buen control metabólico (HbA1c, 5,5 %) con tratamiento dietético. Tiene antecedentes de hemiglosectomía derecha por carcinoma de células escamosas de lengua (libre de enfermedad). Está en seguimiento por Neurología a causa de una TTR-FAP en estadio 2 (mutación V30M), diagnosticada a raíz de haber presentado debilidad en miembros inferiores y ataxia, con electromiograma compatible con una polineuropatía sensitivo-motora de tipo axonal de predominio sensitivo con afectación severa de miembros inferiores. El ecocardiograma no muestra datos de afectación cardíaca por amiloidosis. Desde el punto de vista funcional es totalmente autónomo, pero precisa andador.
En el momento de su primera consulta en Nutrición recibía tratamiento con atorvastatina (10 mg/día), atenolol (25 mg/día) y diflunisal (500 mg/12 h). Refería un peso habitual de 62-63 kg, con pérdida ponderal progresiva en los seis meses previos y disminución de la ingesta. Negaba tener anorexia, síntomas digestivos que dificultasen la ingesta, trastornos del ritmo intestinal, distensión y dolor abdominal. La encuesta dietética mostraba unos hábitos de alimentación equilibrados, con tres comidas principales y dos tomas intermedias, y alimentos de todos los grupos, aunque con tamaños de ración insuficientes. En cuanto a la exploración física, el peso era de 52 kg (pérdida de peso del 15,8 % en seis meses), con un índice de masa corporal (IMC) de 18,7 kg/m2, pérdida de masa muscular y presencia de edema en el dorso de los pies. Aportaba una analítica reciente con una Cr de 1,5 mg/dl (cifras habituales) y una FG de 44 ml/min/1,73 m2, correspondiente a una enfermedad renal crónica (ERC) en estadio 3B (KDIGO). Se le diagnosticó una desnutrición severa asociada a enfermedad según los criterios ESPEN. Se le proporcionó consejo dietético, se le prescribió un suplemento nutricional hipercalórico y normoproteico, y se solicitó un estudio analítico.
En la analítica destacaban: glucosa, 123 mg/dl; HbA1c, 5,5 %; FG, 38 ml/min/1,73 m2; proteínas viscerales, iones, pruebas de función hepática (PFH), perfil lipídico, perfil férrico, función tiroidea, vitamina B12, folatos, vitamina B1y vitamina B6dentro del rango de normalidad; 25-OH D, 17 ng/dl; cobre (Cu), 29,17 mcg/dl (rango normal de 70-140 mcg/dl para el sexo y la edad del paciente); Hb, 13,4 g/dl; Hto, 39,4 %; VCM, 92 fL; leucocitos, 5190 x 103/mcl; y proteínas en orina negativas. Ante el hallazgo de unos niveles bajos de Cu sérico se interrogó al paciente sobre la ingesta de suplementos de zinc (Zn), así como sobre la composición del colutorio y la pasta de fijación de la prótesis dental que utilizaba, descartándose un aporte excesivo de Zn a partir de estas fuentes. Se solicitaron unos niveles de Zn séricos y un estudio del metabolismo del Cu (Tabla I), así como anticuerpos anti-transglutaminasa tisular, anti-péptidos deaminados de gliadina y anti-endomisio, que resultaron negativos. Se realizó una resonancia nuclear magnética (RNM) de médula cérvico-dorsal en la que no se objetivaron alteraciones focales de la señal intramedular a nivel de los cordones posteriores.
Tabla I. Resultados del estudio del metabolismo del cobre y los niveles de zinc
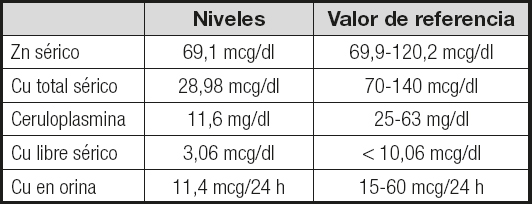
Zn: zinc; Cu: cobre.
El paciente, tras ser informado, rechazó la administración de Cu por vía intravenosa (IV), por lo que después de la recogida de las nuevas muestras se inició el tratamiento por vía oral con 6 mg de Cu diarios. Al cabo de cuatro meses había recuperado su peso habitual y los niveles de Cu sérico permanecían sin cambios. Se aumentó la dosis a 8 mg diarios, sin objetivarse mejoría de la cupremia tras otros cuatro meses. A petición del paciente, dado que no había presentado mejoría de la clínica neurológica ni normalización de los niveles séricos de Cu, se suspendió el tratamiento, estando pendientes de su evolución en el momento de redactar este artículo.
DISCUSIÓN
El Cu es un oligoelemento que funciona como componente de las cuproenzimas. En la tabla IIpueden verse las más importantes y su función fisiológica (1). Nuestro organismo contiene unos 100 mg de Cu, más de la mitad del cual se encuentra en el hueso y el músculo, hallándose las concentraciones más altas en hígado, riñón y cerebro (2). Solo el 5 % está en sangre, el 95 % unido a ceruloplasmina (Cp) y el restante a albúmina y aminoácidos. La dieta normal contiene 1-5 mg de Cu. Su presencia en los alimentos es ubicua, pero son especialmente ricos en Cu las vísceras, la carne, el marisco, los frutos secos, las legumbres, los cereales y el chocolate. Las recomendaciones diarias (RDI) son de 0,9-1,2 mg/día para los adultos, de 1 mg/día durante la gestación y de 1,3 mg/día en la lactancia. El límite superior de tolerabilidad (ULT) es de 10 mg/día. Se absorbe en el estómago, el duodeno y la primera porción del yeyuno entre el 55 y el 75 % del cobre ingerido, dependiendo del aporte dietético, el estado del Cu, el pH luminal (la acidez favorece la absorción) y el efecto de otros nutrientes. La vitamina C, el hierro (Fe) y el zinc (Zn) disminuyen la absorción (2), y los fructoligosacáridos de cadena corta y el déficit de Fe la aumentan. Una vez absorbido se transporta al hígado, donde se almacena y a partir del cual se libera a la circulación sistémica o se excreta en la bilis. El 80 % del Cu excretado está en la bilis y las secreciones gastrointestinales (el 10-15 % se reabsorbe por la circulación enterohepática), y el 20 % en la orina.

En la Tabla IIIse muestran las causas fundamentales del déficit de Cu, adquiridas (3,4) y genéticas (5).
La ATP7A es una molécula necesaria para el transporte del Cu tanto dentro de la célula como para su salida de esta. Las mutaciones a nivel de esta molécula causan tres fenotipos distintos dependiendo del defecto molecular. La enfermedad de Menkes, descrita en 1962 por John Menkes, se presenta entre las seis semanas y el año de vida y cursa con neurodegeneración severa con desmielinización, atrofia cerebral, convulsiones, retraso del crecimiento, hipotonía, hipopigmentación cutánea, cabello hipopigmentado y rizado, aneurismas, defectos cardíacos, etc. Es mortal en la infancia y no responde al tratamiento con Cu. Los niveles de Cu y Cp están bajos. El síndrome del cuerno occipital (OHS) es una variante más leve de la enfermedad de Menkes que se manifiesta entre los 3 y 10 años con laxitud cutánea y articular. Otros hallazgos son los divertículos de vejiga y los síntomas de disautonomía (ortostatismo, bradicardia). Es característica la presencia de exostosis ósea occipital en los estudios radiológicos. Los niveles de Cu y Cp séricos son normales o bajos. En los últimos años se ha descrito una neuropatía motora distal relacionada con ATP7A, causada por una mutación sin sentido. Se manifiesta entre los 5 y los 50 años de edad con debilidad y atrofia lentamente progresiva de los músculos distales y no presenta anomalías bioquímicas específicas (5). La enfermedad de Wilson se debe a una disfunción hereditaria autosómica recesiva de ATP7B, que regula el transporte de Cu a la bilis. Como resultado se altera la excreción de Cu, que se acumula en el hígado (cirrosis), los ganglios basales (clínica neurológica y psiquiátrica) y la córnea (anillo de Kayser-Fleischer). Su diagnóstico se basa en el hallazgo de niveles séricos bajos de Cu y Cp, concentraciones elevadas de Cu en el hígado y excreción elevada de Cu en la orina. Pueden hallarse niveles de Cu sérico altos en los pacientes con fallo hepático, debido a la liberación del Cu acumulado en los hepatocitos (1). En nuestro paciente, el cuadro clínico y el nivel de Cu en orina bajo descartan cualquiera de estos trastornos genéticos.
Nuestro paciente tampoco presenta, aparentemente, ninguna de las causas habituales del déficit de Cu adquirido (Tabla III). Si bien, al inicio, la ingesta de nutrientes resultaba insuficiente, esta aumentó de forma significativa en pocas semanas. Además, el hecho de que los niveles de Cu no mejorasen con la administración de Cu por vía oral haría pensar más en un problema de aumento importante de las necesidades o de las pérdidas, o en un déficit de absorción. La historia clínica nos permite descartar los factores etiológicos que se relacionan con un aumento de las necesidades o las pérdidas de Cu. Por otra parte, no ingiere alcohol ni recibe tratamientos farmacológicos distintos de los descritos, no tiene antecedentes de cirugía digestiva, se ha descartado la sobrecarga de Zn, no presenta sintomatología digestiva y los anticuerpos de celiaquía son negativos. A esto se suma que los niveles de otros micronutrientes habitualmente alterados en los cuadros de malabsorción son normales. Todo ello hace pensar en un defecto específico en la absorción de Cu que desconocemos. La causa del déficit de Cu permanece sin aclarar en el 20 % de los casos (6).
Tabla III. Causas de los niveles de cobre bajos
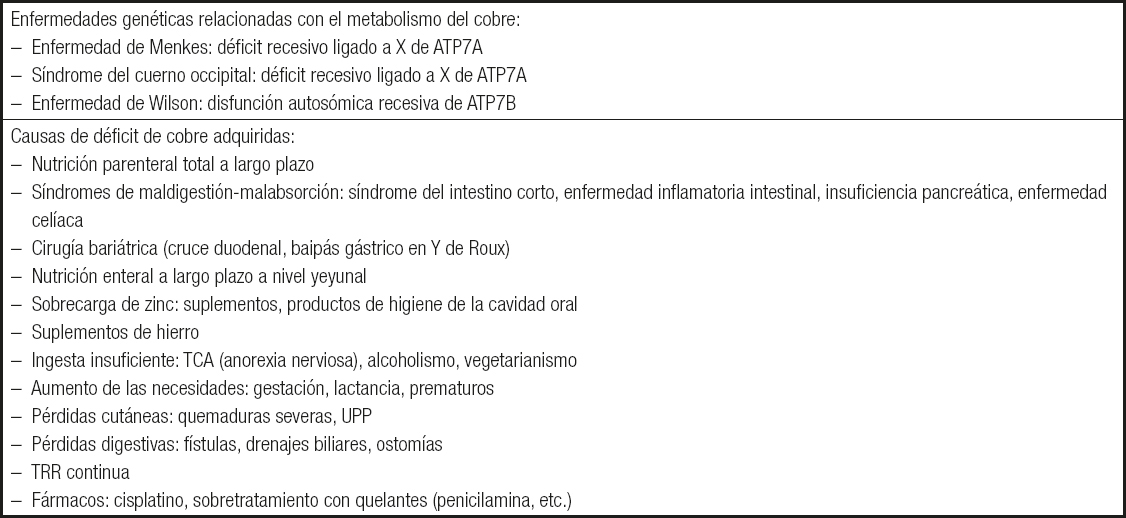
TCA: trastornos del comportamiento alimentario; UPP: úlceras por presión; TRR: terapia de reemplazo renal.
Dado que nuestro paciente presenta una TTR-FAP, se realiza una búsqueda bibliográfica en las bases de datos PubMed, EMBASE, WOS, Scopus y Lilacs con la siguiente estrategia: "copper shortage OR copper deficiency OR copper deficit OR copper deficiency OR hypocupremia AND transthyretin familial amyloidosis OR transthyretin polineuropathy OR amyolid neuropathies, familial OR TTR-FAP OR hereditary transthyretin polineuropathy", sin hallar ningún resultado.
El 69 % de los pacientes con TTR-FAP V30M presentan síntomas gastrointestinales después de una media de 5 años en relación con una neuropatía autonómica y entérica que lleva a una alteración de la motilidad intestinal, con sobrecrecimiento bacteriano y malabsorción de ácidos biliares como complicaciones comunes (7). Los síntomas más habituales son: saciedad precoz, náuseas, vómitos, retención gástrica, estreñimiento, alternancia diarrea-estreñimiento y diarrea constante (con frecuencia en combinación con incontinencia). Con menor frecuencia aparece disfagia. Nuestro paciente no presenta síntomas digestivos. La pérdida de peso es común en los pacientes con TTR-FAP, pudiendo preceder a los trastornos gastrointestinales (8), como ocurre en este caso.
Cuando la ingesta de Cu es inferior a los requerimientos, inicialmente se repone a partir de los depósitos hepáticos, por lo que las manifestaciones clínicas pueden tardar en presentarse. En el caso de la omisión del Cu en la nutrición parenteral (NP) aparecen entre 6 semanas y 15 meses después, y en los pacientes sometidos a cirugía bariátrica pueden tardar hasta 9 años. En la Tabla IVaparecen las manifestaciones clínicas más importantes de la hipocupremia (3,4,9,10). A nivel hematológico, los hallazgos más frecuentes son la anemia microcítica o normocítica y la neutropenia, con una médula ósea que simula un síndrome mielodisplásico. Ante la presencia de anemia resistente al tratamiento con hierro debemos pensar en la posibilidad de una hipocupremia. El cuadro neurológico más común es la mielopatía, similar a la degeneración combinada subaguda medular por déficit de vitamina B12, que se presenta con ataxia sensitiva y trastorno de la marcha. Un 44 % de los pacientes con mielopatía muestran en las imágenes de RNM un incremento en la señal T2 de la columna dorsal de la médula cérvico-torácica (11). Nuestro paciente no presenta hechos hematológicos relevantes, la RNM de médula cérvico-torácica no muestra aumentos de señal y ya había de base una neuropatía periférica en relación con la TTR-FAP.

Los objetivos del tratamiento con Cu en pacientes con déficit son: tratar los síntomas asociados, normalizar las concentraciones de Cu sérico, restaurar los almacenes corporales y evitar las recaídas. Las dosis de Cu óptimas son empíricas. La correlación entre las concentraciones de Cu sérico, los depósitos tisulares y la recuperación neurológica es desconocida; por ello, las dosis deberían individualizarse en función de la respuesta clínica y los cambios en los niveles de Cu y Cp (10). En general, la recuperación hematológica suele ser completa en unas 4 semanas; sin embargo, las manifestaciones neurológicas pueden mejorar solo parcialmente o estabilizarse más que recuperarse de forma completa. La hipocupremia severa en adultos puede tratarse con Cu IV (2-4 mg/día) durante 6 días, seguido de Cu por vía oral (3-8 mg/día) en forma de sulfato o gluconato. En nuestro caso, la administración de Cu por vía oral no mejoró las cifras de Cu sérico y el paciente rechazó la posibilidad de tratamiento IV.
En conclusión, presentamos el caso de un paciente con una hipocupremia severa de etiología no aclarada que no responde al tratamiento vía oral, y en el que desconocemos hasta qué punto podría haber alguna repercusión neurológica. No hemos encontrado bibliografía que relacione la hipocupremia con la TTR-FAP.

