










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCTION
Cobalamin C (Cbl C) deficiency is the most common inborn error of intracellular cobalamin metabolism. It is caused by mutations in the MMACHC gene located on chromosome 1p34 (1). This mutation results in a decrease in intracellular conversion of vitamin B12 to its two metabolically active forms, adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl). These are essential coenzymes to methylmalonyl-CoA mutase and methionine synthase (2). A decrease in the activity of these enzymes causes an increase in methylmalonic acid and homocysteine levels in plasma and urine with a decrease in the production of methionine (3). Age at onset differentiates two main forms of presentation: an early-onset form, with a more severe presentation, generally in the first year of life; and a late-onset form, with a milder, more heterogeneous presentation predominantly affecting the central nervous system (4). Clinical suspicion and early diagnosis are fundamental pillars in order to avoid the progression and irreversibility of this multisystemic disease.
CASE REPORT
In this paper we report the case of a 45-year-old man who was referred to our Inherited Metabolic Diseases clinic in 2017 for assessment of a potential metabolic disease due to elevated homocysteine and methylmalonic acid levels in the blood. The patient had chronic kidney disease (CKD) diagnosed in 1995, which was believed to be caused by chronic pyelonephritis; however, a histological study was never performed. He had been on renal replacement therapy through hemodialysis for 3 years. He had no family history of consanguinity or known metabolic diseases. He denied consumption of alcohol and/or drugs.
As associated complications he had presented with repeated episodes of thrombosis in the arteriovenous fistula (up to 4 times), 4 catheter replacements due to thrombosis, and several episodes of tonic-clonic seizures of unclear etiology during hemodialysis. In addition, there was a history of acute encephalopathy with spastic paraparesis in 2010, which he had presented in the context of acute appendicitis and was initially treated with corticosteroid therapy because of a suspected autoimmune etiology. After treatment he experienced a partial improvement in his encephalopathy, but developed a neurogenic bladder and a worsening of lower limb mobility. In 2012 he presented again with an episode of encephalopathy with similar characteristics. The lower limb weakness progressed, completely limiting ambulation, and he was diagnosed with spastic paraparesis of unknown origin. The magnetic resonance images obtained during the neurological study are provided in figure 1.
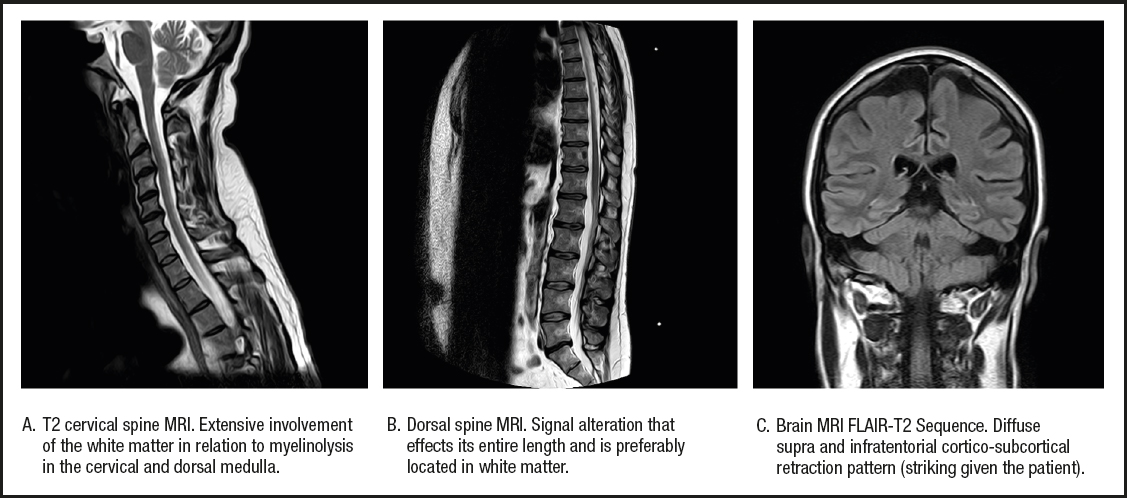
As a consequence, the patient was studied on multiple occasions by nephrologists and neurologists, who observed elevated levels of homocysteine in the blood, as well as blood folic acid and vitamin B12 levels within the normal range. Despite this, treatment was started with folic acid and vitamin B12 (cyanocobalamin) supplementation.
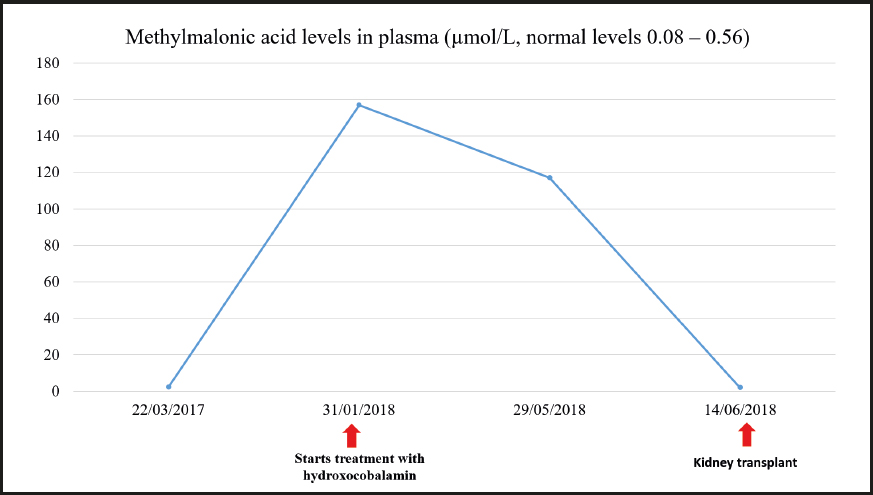
Subsequently, the neurological condition was studied at the Toledo Paraplegic Hospital, Spain, where very high levels of methylmalonic acid — 117.18 µmol/L (normal range, 0.08-0.56 µmol/L) — were detected in the blood. Blood levels of vitamin B12 — 1862 pg/mL (normal range, 220-900 pg/mL) — and folic acid — 20 ng/mL (normal range, 2.5-17 ng/mL) — were also found to be slightly increased. These results prompted physicians to refer the patient to our Inherited Metabolic Diseases clinic.
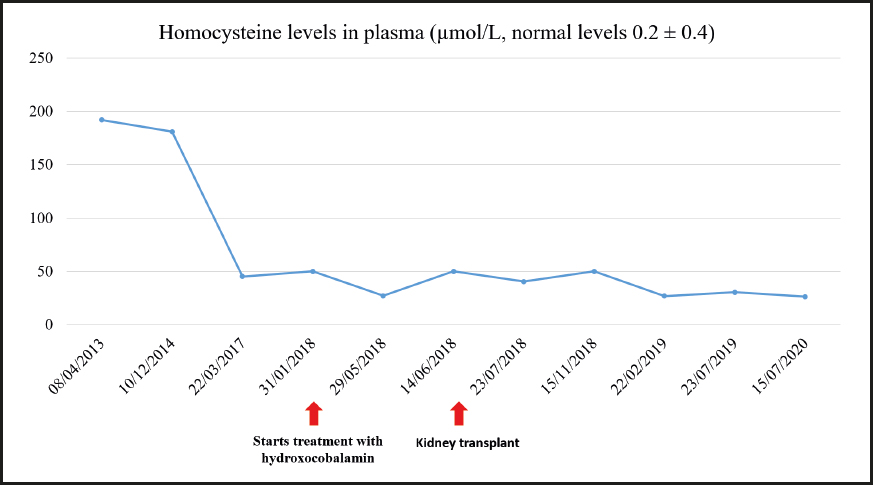
Since the patient presented with anuria due to dialysis, and a urine sample could therefore not be obtained, a larger metabolic study was decided to be carried out using blood chemistry. The levels of methylmalonic acid in the blood were 2.32 µmol/L and those of homocysteine were 45.4 µmol/L. The study was completed with an assessment of pristanic acid, phytanic acid, and very long-chain fatty acids in the serum, which came back normal. A genetic study confirmed methylmalonic acidemia with homocystinuria due to Cbl C deficiency related to mutations in the MMACHC gene. Two variants previously described as pathogenic (p.Ser146Argfs*35 and p.Arg161Gln) were identified in the MMACHC gene. Therefore, a genetic study of the parents was recommended to confirm the presence of variants in different alleles.
Treatment was started with hydroxocobalamin at a dose of 1 mg/day IM initially, which was subsequently down-titrated to twice a week as maintenance dose. Additional treatment consisted of 3 g every 8 hours of betaine, 20-25 mg/kg of carnitine, and 5 mg of folic acid, daily. After starting treatment the patient experienced a partial improvement of his neurological manifestations. He received a kidney transplant in June 2018 once the etiology of CKD was known.
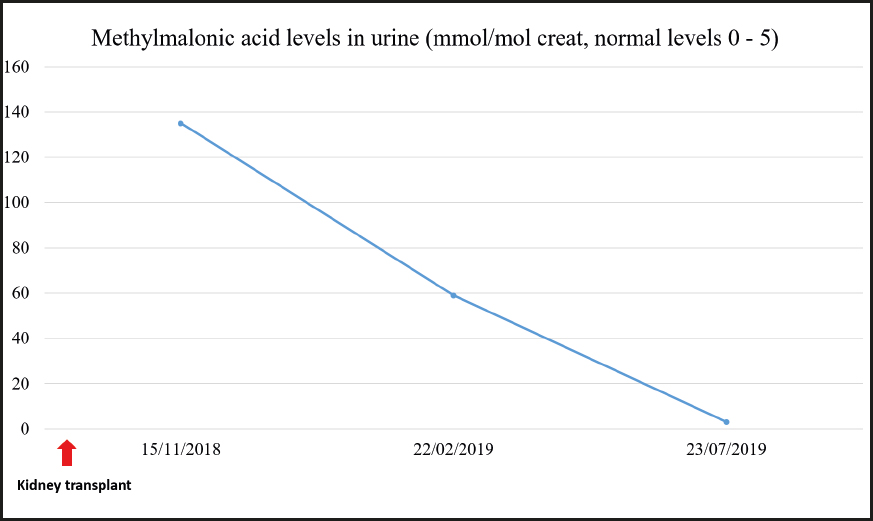
During the last visit in July 2020 the patient showed good metabolic control with methylmalonic acid levels within the normal range, and slightly elevated homocysteine levels. The evolution of homocysteine and methylmalonic acid levels is shown in figures 2, 3 and 4. The patient reported that he was well, maintaining an active life, going to physical rehabilitation and a gym on a regular basis. He needs a wheelchair to move around, although he has regained some mobility in the lower limbs. He is the carrier of a permanent urinary catheter but renal function is preserved and stable.
DISCUSSION
In this paper we report the case of a patient with a rare metabolic disease that because of being associated with late onset, heterogeneity in clinical presentation, and absence of clinical suspicion represents a diagnostic challenge. Although this is a rare disease, in 2007 there were about 250 cases published in the literature (4). However, when focusing exclusively on patients with late presentation, in 2014 Huemer et al. published a review of the literature in which they included 61 cases (5). Subsequently, in 2019 Wang et al. reported 16 new cases (2).
Despite the fact that late presentation represents a less severe form, and has a more favorable prognosis, than early presentation, the diagnosis is usually delayed due to an absence of clinical suspicion. Huemer et al. described that diagnosis may be delayed for more than 20 years, as was the case with our patient (5).
Regarding the clinical symptoms, myelopathy and dementia are the most frequent and invalidating characteristics that these patients face. The study by Wang et al. revealed that up to 100 % of their patients presented with alterations in the pyramidal tract on physical examination (2). Their pathophysiology appears to lie in the demyelination caused by low levels of adenosylcobalamin, in methionine deficiency, and in the neurotoxicity caused by high levels of methylmalonic acid (5).
Although representing a rare clinical feature of this condition (5), kidney disease was the initial manifestation in our patient. The delayed diagnosis probably influenced the progressive deterioration of renal function, and the need for renal replacement therapy. Furthermore, the lack of an accurate diagnosis in this regard meant that the patient was not initially a candidate for kidney transplantation. Finally, after reaching a diagnosis and prescribing an optimal treatment the patient was able to receive a kidney transplant in 2018, and currently has preserved kidney function.
Associated with this entire clinical spectrum is a state of hypercoagulability caused by hyperhomocysteinemia, which manifests as an increased incidence of thrombosis (6). Our patient had suffered from a total of 4 fistular thromboses and 4 catheter thromboses. In this case, elevated levels of homocysteine in the blood were accounted for by renal failure although, usually, when it comes to renal failure, homocysteine levels are not as high as in the case reported herein (7).
It should be noted that, in this pathology, the affectation of vitamin B12 metabolism is intracellular, and therefore plasma levels of vitamin B12 remain normal, which renders the diagnosis extremely difficult. In fact, in our patient, they had been measured on several occasions and the results were always normal.
Therefore, within the initial study of myelopathies, CKD, and thrombophilias of unknown origin it is necessary not only to measure vitamin B12 and folic acid levels in the blood, but also homocysteine, organic acids, and other amino acids both in the urine and plasma (8).
Finally, we believe that the publication of these clinical case reports (including ours) provides scientific knowledge on infrequent causes of nephrological and neurological disorders, in which an accurate diagnosis and early treatment are a priority to improve the progressive and disabling course of disease, with the potential to even reverse it when in early stages (8).
CONCLUSION
The establishment of appropriate treatment considerably improves the clinical outcome of patients with Cbl C deficiency. A measurement of homocysteine, organic acid, and other amino acid levels should be included in the differential diagnosis of patients with nephrological-neurological symptoms of unclear etiology.

