










 Custom services
Custom services
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Autophagy is the term (derived from the Greek words "auto" meaning "oneself" and "phagein" meaning "eating") adopted to describe the set of molecular reactions that result in the degradation of intracellular components in lysosomes, a process for which Yoshinori Ohsumi was awarded the Nobel Prize in Physiology or Medicine in 2016 (1). Recycling is one of the bases of cell survival, since it allows to reduce cellular waste, preserve cellular energy, and adapt to different changes by regulating the abundance of intracellular components. Autophagy occurs in cases of low basal metabolic rate (activated in starvation and inhibited in the presence of nutrients) and is elevated during differentiation and remodelling in a wide variety of tissues, such as brain, intestine, kidney, adipose tissue, lung, liver, prostate, skin, thyroid gland (1), also in the control of cell growth and defence (2) and in the prevention and triggering of some pathologies (3). Contrary to what was previously believed, autophagy is not only based on a protein recycling process, but also contributes to maintaining a positive energy balance through the degradation and use of organelles, glycogen or lipids.
TYPES OF AUTOPHAGY
Based on the way substrates reach the lysosomal lumen, three main forms of autophagy have been described in mammalian cells: macroautophagy, chaperone-mediated autophagy, and microautophagy (1,4) (Fig. 1).
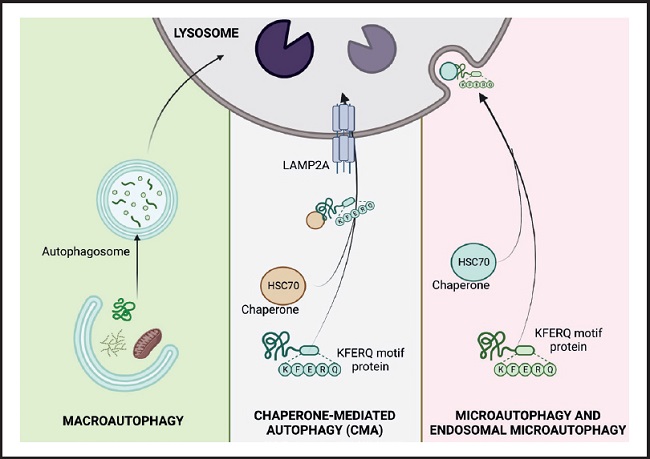
Figure 1. The three types of autophagy in mammalian cells. Macroautophagy relies on de novo formation of cytosolic double-membrane vesicles, autophagosomes, to sequester and transport cargo to the lysosome. Microautophagy involves the direct uptake of cargo through invagination of the lysosomal membrane. Chaperone-mediated autophagy (CMA) transports individual unfolded proteins directly across the lysosomal membrane. All three types of autophagy lead to degradation of cargo and release of the breakdown products back into the cytosol for reuse by the cell. (HSC 70: cytosolic heat shock-related protein of 70 kDa; KFERQ: pentapeptide domain. Modified from Macho-González and Sánchez-Muniz (35).
MACROAUTOPHAGY
This is the best characterized lysosomal recycling pathway, in which cytosolic elements destined for degradation are sequestered by the phagophore, a double-membrane vesicle of non-lysosomal origin, which delivers the cytosolic material to lysosomes. The phagophore is formed through the assembly of proteins and lipids from different organelles such as the endoplasmic reticulum, Golgi apparatus or plasma membrane. The closure of the vesicle with the cargo inside gives rise to what is known as an autophagic vacuole or autophagosome, which moves through the microtubules to fuse with the lysosomes, organelles that provide the enzymes necessary for the degradation of the components sequestered (5,6).
CHAPERONE-MEDIATED AUTOPHAGY
This was discovered by J. Fred Dice in 1985 and is an autophagic pathway for the selective degradation of proteins that cross the lysosomal membrane through the membrane protein associated with the lysosome type 2A (LAMP-2A) (4). In this process, cytosolic proteins destined for degradation must be recognized by the chaperone cytosolic heat shock-related protein of 70 kDa (HSC 70) through a pentapeptide domain (KFERQ) of the protein's amino acid sequence. Once the chaperone-substrate complex reaches the lysosomal membrane and binds to monomeric LAMP-2A, its multimerization is induced and the substrate protein is transported into the lysosome for degradation. The activation of this type of autophagy, in addition to acting in response to nutritional changes, also functions as a defence mechanism against cellular attacks to eliminate damaged proteins and ensure adequate protein homeostasis (2,4).
MICROAUTOPHAGY
In this type of autophagy the loads are sequestered by direct invagination or protrusion of the lysosome, originating small single-walled vesicles from the lysosomal membrane (7). Recently, a variation of this process called endosomal microautophagy has been found, which takes place in late endosomes and involves selective degradation through recognition of the KFERQ domain of proteins by hsc70, as occurs in CMA. However, substrate internalization is via the transport-required endosomal sorting complex I and III (ESCRTI and III) rather than LAMP-2A.
CENTRAL MOLECULAR ASPECTS OF AUTOPHAGY
The autophagy process can be divided into phases: 1) nucleation, 2) elongation, 3) formation of the mature autophagosome, 4) fusion, 5) degradation, and 6) recycling. The nucleation phase is regulated by the mTORC1 complex, whose regulation depends on the AMP/ATP ratio which, in turn, is related to nutritional and metabolic status. This complex and downstream proteins are regulated by phosphorylation/dephosphorylation. A large part of the process is mediated by proteins of the ATG family (autophagy-initiating proteins), which bind to damaged proteins or organelles, marking them for the formation of phagophores (double-membrane vesicles that contain the material to be recycled). Some ATG proteins bind to phagophores, promoting their maturation and fusion with the lysosome to form the autophagosome (8) (Fig. 2).
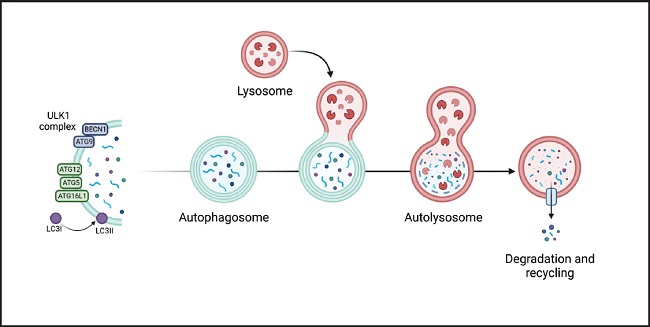
Figure 2. Genesis and development of the autophagosome and participation of the different ATG proteins in its 6 stages (nucleation, elongation, formation of the mature autophagosome, fusion, degradation, and recycling. It begins by activation of the ULK1/2 kinase-kinase complex (proteins uridine type kinase 1 and 2, respectively), which phosphorylates the Beclin1 protein (BECN1, also known as ATG6) that is part of the phagophore nucleation initiator complex and subsequently its conjugation with the ATG5/ATG12/ATG16 complex (initiator proteins 5, 12 and 16). It, then, continues with the processing of LC3 (microtubule-associated protein light chain 3) and the insertion of the phagophore into the membrane producing the formation of the autophagosome which then fuses with the lysosome (autophagolysosome), where the degradation of the substrate occurs. Finally, degradation products are released into the cytoplasm to be eliminated or recycled. Modified from Costas and Rubio (34).
The presence of ATG proteins is essential for a proper functioning of the cell recycling process, which has been observed to play roles of indisputable importance in health, and alterations in the autophagic process are implicated in the pathophysiology of cardiovascular diseases, infectious diseases, Crohn's disease and neurodegenerative disorders (9,10).
DIET AND BIOACTIVE DIETARY COMPOUNDS AS MODULATORS OF AUTOPHAGY
Starvation was the first stimulus described to activate the main autophagic pathways (1,2). In the absence of nutrients, autophagy promotes the breakdown of intracellular proteins to ensure the necessary amino acids for protein synthesis. In turn, they also use these pathways to mobilize intracellular lipid and glycogen stores to generate energy when nutrients are scarce.
The communication between nutritional status and autophagy is mediated by intracellular nutrient sensors, the main player of which is the mammalian target of rapamycin (mTOR). This kinase integrates nutritional signals (amino acids, insulin, and ATP levels) and actively suppresses macroautophagy under normal nutritional conditions (11). However, nutrient deprivation increases the AMP/ATP ratio, which activates AMP-activated protein kinase (AMPK), which promotes autophagy and autophagosome formation by inhibiting mTOR and phosphorylating uridin-like-kinase 1 (ULK1). It is well known that mTOR and AMPK nutritional sensors can be modulated by different food components such as polyphenols. It has been suggested that a diet rich in polyphenols can protect against aging by inducing autophagic activity, being one of the mechanisms underlying the beneficial properties attributed to them and favouring the recycling of cellular waste (12). Quercetin and resveratrol increase lifespan in lower eukaryotes by activating sirtuin (SIRT1), a deacetylase that induces autophagy through deacetylation of autophagic genes and its interaction with AMPK (13). However, not all polyphenols show the same action, nor are their bioavailability the same, nor are the effects greater at high concentrations. Polyphenols are showing their therapeutic potential in diseases that occur with alterations in autophagy pathways, since they can modulate key points in degradative processes such as mTOR, AMPK, p62/SQSTM1 or Nrf2. Although the molecular mechanisms involved are still unknown, these compounds represent a promising nutritional strategy (14).
ROLE OF ATG GENE POLYMORPHISMS IN PATHOLOGIES
Since the discovery of genes related to autophagy in yeasts, it is known that the correct functioning of autophagy depends on the presence of polymorphisms in ATG genes, being possible to speak of candidate genes to be studied primarily in errors in autophagy for the ATG 5,12 and ATG16L-1 genes (5,15). The presence of the ATG protein that intervenes in the formation of the phagophore depends on the expression of the ATG genes.
Therefore, the fight against many degenerative diseases, including different types of tumours or cancer, begins with knowledge of the autophagic transcriptome (a set of mRNAs that originate from DNA and are going to be translated on ribosomes), giving rise to the production of a set of proteins (proteome). Unquestionably, gene expression is also modulated by epigenetic effects. In fact, epigenetic alternations of microRNAs and DNA methylation is using for disease diagnosis and treatment (16,17).
Very interesting promising results on gene epigenome and the Mediterranean diet have been recently published, as a higher adherence to the Mediterranean diet is associated with differential DNA-methylation on relevant autophagy related genes and may have a potential effect upregulating autophagy (18).
Discoveries about autophagy have laid the groundwork for knowing its physiological involvement in a variety of physiological and pathological states, having established relationships with Alzheimer's disease, cancer, obesity, T2DM or non-alcoholic fatty liver disease (2,6,15).
ROLE OF AUTOPHAGY IN TYPE-2 DIABETES MELLITUS
Insulin resistance in peripheral tissues and decreased insulin production by pancreatic β-cells are the two main diabetogenic factors (19). At the beginning of T2DM, pancreatic β-cells adapt themselves to the situation of insulin resistance by increasing the production and secretion of said hormone. This constant demand has been associated with a greater accumulation of misfolded proteins, especially proinsulin, which cause endoplasmic reticulum stress (20). The continuous glucose uptake by the pancreas and its subsequent metabolism induces a high amount of reactive oxygen species that cannot be counteracted by the antioxidant machinery. Likewise, the arrival of free fatty acids and proinflammatory cytokines from peripheral tissues also contributes to and aggravates cellular stress, causing pancreatic β-cell dysfunction and failure (20). Autophagy can prevent cellular stress by removing misfolded proteins and non-functional organelles such as damaged mitochondria. However, in the diabe- tic situation, autophagic activity may be reduced or altered though stimulation of mTORC1 by glucose and free fatty acids or though inhibition of lysosomal acidification by free fatty acids and amylin accumulation, leading to destruction of β-cells and contributes to the vicious cycle of T2DM (21).
On the other hand, mitochondria play a central role on cellular metabolic homeostasis; in such a way that the maintenance of a healthy mitochondrial population is essential for cell survival. Metabolic overload of mitochondria clearly influences both muscle insulin resistance and β-cell dysfunction. This overload leads to incomplete β-oxidation, oxidative stress, accumulation of toxic lipid intermediates, and mitochondrial damage (22). Mitophagy, a macroautophagy type consisting in the elimination of aged and altered mitochondria, has been defined to play a protective role against development of insulin resistance and diabetes, as it eliminates oxidative stress and its related pathogenic processes (23). Autophagy also mediates exercise-induced increases in muscle glucose uptake and protects β-cells against ER stress in diabetogenic conditions (21). On the other hand, adipose tissue autophagy promotes adipocyte differentiation, possibly through its role in mitochondrial clearance. Being involved in many aspects, autophagy appears to be an attractive target for therapeutic interventions against obesity and diabetes (22,23).
T2DM is characterized by mitochondrial dysfunction, high production of reactive oxygen species (ROS), and low levels of ATP (24). Dynamic processes such as mitophagy and mitochondrial biogenesis are also clearly affected in T2DM (24).
As previously discussed, autophagy is clearly dependent of the phagophore formation, whose constituents' proteins are in turn expressed by autophagy genes. Most studies indicate that alterations in the liver, pancreas and adipose tissue participate in T2DM pathophysiology. These alterations are related to modifications in the autophagic process. Thus, knockout mice for Atg7, a gene essential for macroautophagy activity as we have previously discussed, showed structural and functional defects in pancreatic β-cells, leading to glucose intolerance (25). This same deletion in the liver resulted in liver hypertrophy and triglyceride accumulation because of a lipophagy dysregulation, while a lower fat deposition, due to a defective differentiation of adipocytes, is induced in adipose tissue (20).
Similar results have been found for hepatic LAMP-2A knockout mice. These animals presented a phenotype that manifested itself with very high rates of glycolysis and reduced neoglycogenesis and lipolysis, leading to the development of hepatosteatosis (26).
Despite all the indications that associate an alteration of autophagy with T2DM, additional studies are required to know if deregulation of autophagic activity is a cause or consequence of said pathology.
Considering the previous premise, briefly mentioned in this review, there is no doubt that autophagy is a very important new aspect to take into account in the diagnosis, treatment and resolution of very prevalent degenerative conditions. In the very near future, the population should be genotyped according to genes engaged in the encoding of autophagy central proteins (e.g., ATG proteins, necessary for the formation of the phagophore), as a great variability in response to diet has been defined between some individuals and others due to polymorphisms in candidate genes. This will allow to perform a precision nutrition to ensure the greatest possible success in all individuals given the known differences in response between subjects (27).
TYPE-2 DIABETES MELLITUS DRUGS AND AUTOPHAGY
Present evidence suggests that antidiabetic agents can affect autophagy pathways and modulate this process. In this section the role of metformin, SGLT2i and DPP-4 inhibitors and GLP-1 receptor agonists (GLP-1RA) on autophagy is reviewed. Metformin hydrochloride is the most common oral glucose-lowering medication. The guidelines published by national and international diabetes associations have supported the use of this drug as first-line therapy together with lifestyle changes for the treatment of adults with newly diagnosed T2DM with or without risk of cardiovascular disease. This reputation has resulted from its effective glucose-lowering capabilities, low cost, weight neutrality, and good overall safety profile (especially the lack of hypoglycemia as an adverse effect) (28-30).
GLP-1RA drugs (e.g., liraglutide) have proven to be highly effective in the treatment of diabetes (31). GLP-1RA result in pharmacological levels of glucagon-like peptide-1 (GLP-1), an incretin produced and secreted by the intestinal enteroendocrine L-cells that can decrease blood sugar levels in a glucose-dependent manner by enhancing the secretion of insulin (31). The DPP-4i inhibitor class drugs (e.g., omarigliptin) have the function of maintaining endogenous levels of GLP-1 by preventing its breakdown and inactivation (32). Sodium glucose cotransporter 2 inhibitors (SGLT2is) (e.g., gliflozins or flozins) help decrease diabetes hyperglycemia by inhibiting glucose reabsorption in the nephron (33).
AMP-activated protein kinase (AMPK) is a key regulator of numerous pathways and metabolic processes, including glucose metabolism, lipid metabolism, and energetic homeostasis, promoting catabolic reactions that generate more ATP and inhibit energy-consuming anabolic pathways, including gluconeogenesis. AMPK is known to be activated by the knockdown of cellular energy status. In addition, the mammalian target of rapamycin (mTOR) is a key component of the so called mTOR complex 1 (mTORC1) that has been found to be a key regulator of metabolism and autophagy. mTORC1 is inhibited in acute response to depletion of cellular energy (34) (Fig. 3). Although mTOR is tightly controlled under physiological conditions, loss of the negative regulation of this pathway can lead to T2DM (35).
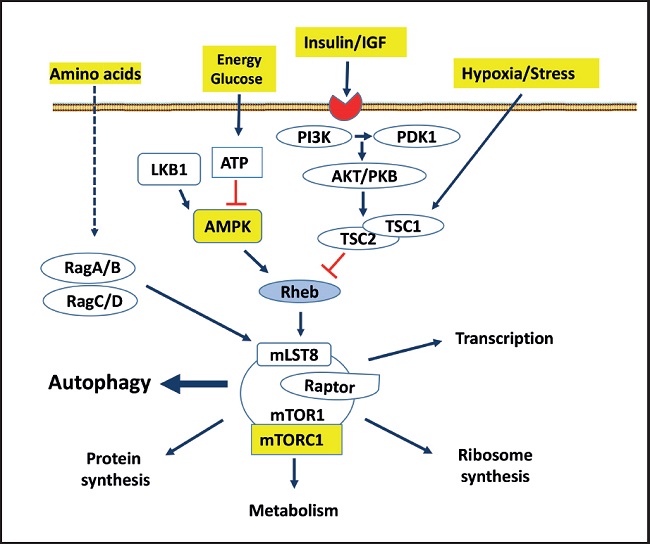
Figure 3. The rapamycin-sensitive mTORC1 pathway controls several pathways, among them autophagy, that collectively determine cell survive and organ health. mTORC1 responds to growth factors (insulin/IGF), the energy status of cells, nutrients (amino acids) and stress. mTORC1, composed of different proteins, is multimeric, although it is schematized as a monomer. The arrows represent activation, while the bars represent inhibition. Dotted lines represent not totally demonstrated or indirect action (PI3K: phosphatidylinositol 3 kinase; Akt/PKB: protein kinase B; PDK: phosphatidylinositol-dependent protein kinase 1; TSC: tuberous sclerosis complex 1 and 2; LKB1: serine threonine kinase encoded by the LKB1 gene; AMPK: adenosine monophosphate-dependent kinase); Rag: Ras-related small GTPases; Rheb: RAS homologue enriched in brain, a GTPase; mLST8: mammalian lethal protein 8 with SEC13; RAPTOR: regulatory-associated protein of mTOR. Modified from Macho-González and Sánchez-Muniz (35).
Metformin exerts its antihyperglycemic effects through stimulating autophagy in a concentration-dependent manner, mainly by suppressing liver glucose production through AMPK-dependent and AMPK-independent pathways (36). Also, AMPK activation leads to mTORC1 inhibition, which also results in suppression of gluconeogenesis (37).
Autophagy can also be affected by novel antidiabetic agents, including GLP-1RAs, DPP-4is and SGLT2is (38). GLP-1RA may be able to modulate the autophagy process (36,39). Mechanistically, it has been reported that these protective effects are mediated by autophagy stimulation via induction of Beclin-1, microtubule-associated protein light chain 3 (LC-3) II, AMPK, PI3K and Akt and inhibition of mTOR (38) (Fig. 2).
Exenatide (a mimetic of GLP-1) effectively reduced fasting glycemia, cholesterolemia, and triglyceridemia, as well as plasma malondialdehyde levels (40). The increased formation of autophagosomes, and the up-regulation of Beclin-1 and LC3A/B-II/I found, suggested that those metabolic protective results due to GLP-1RA were mediated by the induction of autophagy (40).
Animal models have evidenced that several positive effects of DPP-4i (e.g., sitagliptin, vildagliptin) are produced through autophagy induction (41-45). Results appear primarily related to the effect of DPP-4is on GLP-1 levels, which in turn allow adequate insulin release (46). In fact, the positive effects of sitagliptin on hepatic insulin resistance observed in ob/ob mice with hepatic steatosis and insulin resistance were mainly mediated by autophagy activation through the inhibition of the mTOR signaling pathway and stimulation of AMPK (43). Finally, the present evidence suggests that SGLT2i inhibitors (e.g., empaglyfloxin) are able to modulate the autophagy process to contribute to their anti-diabetic effects (36,47,48). Thus, the effect of empagliflozin on mitochondrial function was tested in diabetic rats that had suffered myocardial infarction (49). Empagliflozin, through promoting autophagy, restored the number and size of the mitochondria observed in this animal model (48).
CONCLUSIONS AND FUTURE REMARKS
In recent decades several molecular mechanisms of autophagy and their physiological significance have been revealed, with special mention of the studies that led to the Nobel Prize in 2016 and those carried out by other groups. Degradation is a fundamental function of the cell for the maintenance of life. It is necessary to deepen the knowledge of the dynamics of the membranes that constitute autophagy and advance in the development of systems that allow visualizing the dynamic machinery of autophagy with high spatiotemporal resolution. The identification, by Ohsumi, of the ATG genes in yeast represented an enormous advance in the knowledge of autophagy; however, much remains to be done as we are still in the initial stages of understanding this process. More information is needed on the specific role that ATG gene polymorphisms play on autophagosome membrane formation, as well as on the role of the heat shock protein family A member 8 (HSPA8) gene polymorphisms on encoding the central chaperone molecule Hsc70 (Fig. 1). As major evidence on the effects of drug therapy on autophagy came from animal models, it is urgently demanded that studies be carried out in T2DM patients. Finally, considering the role of nutritional status and antidiabetic drugs on autophagy, and the great variability in response to dietary and pharmacological treatments, we think that in the very near future the population at risk for T2DM should be genotyped according to the genes engaged in the three types of autophagy. This will allow to perform a precision nutrition and pharmacotherapy that will ensure the greatest possible success in all individuals given the known differences in response between subjects. It is also important to note that, under conditions such as stress, autophagy increases to maintain cell survival, as an essential cytoprotective response. Alterations in the autophagic process are implicated in the pathophysiology of several degenerative diseases as T2DM. Autophagy is now recognized as a fundamental process of cell physiology with important implications for health and disease, but the causality or casualty of autophagy on health is still unknown.

