










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.18 no.11 nov. 2001
NOTA CLÍNICA
Apoplejía pituitaria: análisis retrospectivo
en 9 pacientes con adenomas hipofisarios
F. Carral San Laureano, I. Gavilán Villarejo, G. Olveira Fuster, J. Ortego Rojo, M. Aguilar Diosdado
Sección de Endocrinología. Hospital Universitario Puerta del Mar. Cádiz
RESUMEN
La apoplejía hipofisaria (AH) es un síndrome clínico agudo causado por un infarto isquémico o hemorrágico de la glándula hipofisaria, lo cual ocurre casi invariablemente en presencia de un adenoma hipofisario. Aunque pueden presentarse hemorragias intratumorales en ausencia de síntomas en el 9,5 al 25% de los casos, varias series sugieren una incidencia de apoplejías clínicas en el 2% al 10% de los adenomas.
En un estudio retrospectivo desde 1988 a 1998 en 110 pacientes con adenomas hipofisarios, se han objetivado 9 casos de apoplejía pituitaria, presentando una incidencia acumulada del 8,2%. La edad media de presentación fue de 52,4 ± 12,8 años, con una razón varón:mujer de 7:2. Los síntomas observados fueron cefaleas (89%), deterioro visual brusco (78%), vómitos (78%) y paresia de pares craneales oculomotores (33%). El diagnóstico de apoplejía pituitaria fue realizado mediante TAC craneal, presentando todos los pacientes un macroadenoma hipofisario con sangrado intratumoral.
Cinco pacientes fueron intervenidos mediante cirugía transesfenoidal. En el 75% de los pacientes afectados (3/4) se observó mejoría de las alteraciones visuales y en el 100% (3/3) recuperación de la paresia oculomotora. Cuatro pacientes fueron tratados de forma conservadora con esteroides. Los dos pacientes que presentaron alteraciones visuales al diagnóstico, recuperaron completamente sus deficiencias. Durante el seguimiento se objetivó la resolución del adenoma hipofisario por la apoplejía en dos casos, estabilidad tumoral en otro y recurrencia del sangrado al sexto año en el cuarto paciente.
El 62,5% de los pacientes han requerido durante el seguimiento terapia hormonal sustitutiva con hormona tiroidea y esteroides.
PALABRAS CLAVE: Apoplejía hipofisaría. Adenomas hipofisarios. Cirugía transesfenoidal.
Pituitary apoplexy: retrospective analysis of 9 patients with hypophyseal adenomas
ABSTRACT
Pituitary apoplexy is an acute hemorrhage or ischemia infarction of the pituitary gland, almost invariably occurring in the presence of an pituitary adenoma. Although intratumoral bleeding occur in about 9,5 to 25% of pituitary adenomas, various series suggest that clinical apoplexy may be diagnosed in about 2% to 10% of the adenomas.
In a retrospective study from 1988 to 1998 of 110 patients with hypophyseal adenomas, there were 9 cases with pituitary apoplexy, yielding an incidence of 8,2%. Their mean age was 52,4 ± 12,8 años years, with a male to female ratio of 7:2. Symptoms observed were headache (89%), sudden visual deterioration (78%), vomiting (78%) and oculomotor nerves paresis (33%). The diagnosis of pituitary apoplexy was established by computerized tomographic scans, and hypophyseal macroadenoma with intratumoral bleeding was observed in every patient.
Five patients underwent transphenoidal surgery. Improvement of visual deficit was observed in 3/4 (75%) and ocular paresis in 3/3 (100%) of affected patiens.
Four patients were treated conservatively with steroids. Two patiens who had visual deficit recovered it completely without surgery. Two hypophyseal adenomas were resolved spontaneosly after bleeding, one stayed unchanged and another presented recurrence of bleeding at six years of follow-up.
Steroid and thyroid hormone replacement therapy was required in 62,5% of patients.
KEY WORDS: Pituitary apoplexy. Pituitary adenomas transphenoidad surgers.
Trabajo aceptado: 24 de Enero de 2000
Correspondencia: F. Carral San Laureano. Hospital Puerta del Mar. Sección de Endocrinología. Ana de Viya, 21. 11009 Cádiz.
INTRODUCCIÓN
La apoplejía hipofisaria (AH) es un síndrome clínico poco común causado por la expansión brusca de la hipófisis secundaria a un infarto isquémico o hemorrágico. La hemorragia hipofisaria ocurre casi invariablemente en presencia de un adenoma hipofisario, mientras que el infarto isquémico puede ocurrir espontáneamente, en el contexto de un adenoma hipofisario o tras una hemorragia obstétrica (Síndrome de Sheehan) (1-5). Aunque pueden presentarse hemorragias hipofisarias en ausencia de síntomas clínicos o bien con un curso subagudo, el término apoplejía pituitaria se reserva para designar a un cuadro neurológico agudo cuyos principales síntomas son la cefalea intensa y las alteraciones visuales. La incidencia de apoplejías pituitarias sobre adenomas (basado en datos clínicos y radiológicos) en series de la literatura varía entre el 0,6 al 10,5% de los adenomas hipofisarios (1-4). Aunque la urgente descompresión quirúrgica del contenido selar ha sido considerada la actitud más aceptada hasta la actualidad, algunos autores recomiendan una actitud conservadora en determinadas situaciones, debido a que la morbimortalidad de estos pacientes, una vez diagnosticados y tratados, suele ser baja (1,3,6). En el presente estudio hemos analizado los episodios de apoplejía sobre adenomas hipofisarios presentados en nuestro hospital en los últimos 10 años, haciendo especial énfasis en las características clínicas y radiológicas de presentación, factores desencadenantes, tratamiento realizado y evolución posterior en función de la actitud terapéutica adoptada (quirúrgica o conservadora).
CASOS APORTADOS
Se han estudiado de forma retrospectiva los pacientes con adenomas hipofisarios ingresados en nuestro hospital en el período 1988-1998, seleccionándose aquellos casos que presentaron episodios clínicos de cefalea, alteraciones visuales y/o oftalmoplejía, asociado a la presencia de signos radiológicos (mediante TAC o RNM) de sangrado tumoral reciente. Se excluyeron aquellos casos de hemorragias hipofisarias subclínicas descubiertas en estudios radiológicos de rutina o en el acto quirúrgico. El diagnóstico de apoplejía sobre adenoma de hipófisis se confirmó mediante anatomía patológica en aquellos pacientes intervenidos.
Se realizó tanto al inicio como en el transcurso del seguimiento: a) Estudio neurooftalmológico. b) Estudio radiológico mediante radiografía simple y TAC craneal. En la mayoría de los casos se cumplimentó con RNM hipofisaria antes y después de la inyección de gadolinio, tanto en secuencias ponderadas en T1 como en T2. c) Estudio hormonal basal consistente en la determinación de gonadotropinas, esteroides sexuales, TSH y T4 libre, prolactina y cortisol. El eje somatotropo (GH-IGF1) y diversos estudios hormonales dinámicos fueron realizados en algunos pacientes. d) En aquellos pacientes intervenidos se realizó estudio anatomopatológico de la pieza mediante técnicas convencionales. El estudio inmunohistoquímico se efectuó tras la inclusión en parafina y tinción con hematoxilina-eosina, a la que se añadieron sueros primarios anti-GH, anti-PRL, anti-TSH, anti-PRL, anti-TSH, anti-ACTH, anti-LH, anti-FSH y anti-Cromogranina A.
En el período de 1988-1998 se presentaron en nuestro hospital 10 episodios de apoplejía hipofisaria, incluyendo dos eventos presentados en el mismo paciente. Sólo el primer episodio de apoplejía hipofisaria fue incluido en nuestro análisis. En este tiempo fueron diagnosticados 110 adenomas, siendo por tanto la incidencia acumulada de apoplejías hipofisarias del 8,2%. La edad media en el momento de la presentación fue de 52,4±12,8 años, con un rango entre los 27 y los 67 años. La razón varón-mujer fue de 7:2. El seguimiento medio de los pacientes con AH ha sido de 3,5±2,7 años.
Todos los pacientes fueron atendidos e ingresados desde Urgencias por presentar sintomatología neurológica aguda. La cefalea constituyó el síntoma de presentación más frecuente (89%), estando presente en todos menos en un paciente. El 78% de los pacientes presentaron nauseas, vómitos y alteraciones visuales (deterioro brusco de la agudeza y/o campo visual). Cuatro pacientes (44%) presentaron afectación de pares craneales (Tabla I). Posibles factores precipitantes fueron identificados en cuatro pacientes. En dos casos el comienzo de los síntomas coincidió con una crisis hipertensiva, en el tercero la presentación clínica se produjo tras un cuadro de vómitos en el contexto de un cólico biliar y en el cuarto paciente el episodio se presentó 24 horas después de un traumatismo craneal.

Estudios complementarios
Se realizó TAC Craneal de urgencias en 89% de los pacientes (8 de 9) (Figs. 1 y 2), comprobándose la existencia de sangrado en región sellar en el 75% (6 de 8) y presencia de tumoración hipofisaria en el 62,5% de los casos (5 de 8). En el resto de pacientes el diagnóstico de apoplejía sobre adenoma hipofisario se realizó mediante RMN hipofisaria realizada en días posteriores al evento. En el estudio hormonal se evidenció la presencia de normofunción hipofisaria en 4 pacientes (44%), panhipopituitarismo en tres (33%) y déficit aislado de gonadotropinas en 1 paciente (11%). En ningún caso se objetivó hipersecreción hormonal o diabetes insípida.
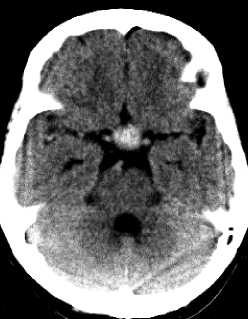

Figs.1 y 2 TAC Craneal al diagnóstico de los pacientes 2 y 3. En ambos casos se aprecia imagen hiperdensa en región sellar compatible con apoplejía pituitaria.
Actitud terapéutica y evolución posterior
Todos los pacientes recibieron terapia con corticoides a elevadas dosis en la fase aguda. Se realizó descompresión neuroquirúrgica urgente en sólo un caso (paciente 8), debido a la severidad de la presentación clínica. En el resto de los casos la actitud terapéutica adoptada (neuroquirúrgica o conservadora) estuvo determinada por la evolución clínica al tratamiento con corticoides. En la tabla II se exponen las principales características evolutivas de todos los pacientes.

Cuatro pacientes (44%) fueron tratados de forma conservadora (altas dosis de corticoides durante la fase aguda) y seguidos durante un tiempo medio de 4,25±2,06 años (2-6 años). Uno de los pacientes presentó recidiva de la AH al sexto año de seguimiento, motivo por el cual fue intervenido. Dos pacientes (50%) tenían un panhipopituitarismo al diagnóstico (persistente hasta la actualidad), mientras que en los otros dos casos han mantenido normofunción hipofisaria durante el seguimiento. Las alteraciones visuales presentes en los dos pacientes afectados evolucionaron favorablemente (recuperación completa de campo y agudeza visual). El estudio radiológico evolutivo ha demostrado la desaparición tumoral en dos casos (hipófisis normal y silla turca vacía), estabilidad del tamaño tumoral en otro y recidiva del sangrado al sexto año de seguimiento en el cuarto paciente.
Cuatro pacientes con AH intervenidos han sido seguidos durante un tiempo medio de 2,85±3,47 años (6 meses a 8 años). En un caso se perdió el seguimiento tras la cirugía. En el 75% de los pacientes (3 de 4) se evidencia en la actualidad un panhipopituitarismo (presente al diagnóstico en un caso). El 100% de los pacientes con afectación de pares craneales recuperaron la normal motilidad ocular en el postoperatorio. Tres de los cuatro pacientes con alteraciones visuales al diagnóstico, mostraron durante el seguimiento estudio campimétrico y de agudeza visual dentro de la normalidad. No existieron complicaciones quirúrgicas significativas. La anatomía patológica fue compatible con adenoma con hemorragia en tres pacientes y adenoma con material necrótico en otros dos, siendo posible realizar estudio inmunohistoquímico en sólo una de las muestras, la cuál fue positiva para tumor secretor de Cromogranina A.
DISCUSIÓN
Se han revisado los casos de apoplejía hipofisaria acontecidos en nuestro hospital con objeto de establecer su incidencia, forma de presentación clínico-radiológica y evolución posterior en función de la actitud terapéutica adoptada.
A pesar de no haber incluido los casos clínicamente silentes, la incidencia acumulada de apoplejía sobre adenomas hipofisarios en nuestra serie es elevada (8,2%) comparada con las descritas en la literatura que oscilan entre el 0,6 al 10,5% (1-4). La edad media de presentación fue de 52,4±12,8 años, dato acorde con la mayoría de las series, donde oscila entre 37,7 y 56,6 años (3-7). Un predominio de hombres sobre mujeres es común en la mayoría de las trabajos (3-7), siendo en la nuestra tres veces más frecuente en varones (ratio 7:2).
La sintomatología predominante en casos de comienzo abrupto de síntomas depende de las estructuras paraselares afectadas, consistiendo el cuadro clínico típico en cefalea de inicio agudo, vómitos, signos de afectación meníngea, disminución de la agudeza y/o campo visual, oftalmoplejía, disminución del nivel de conciencia y, en ocasiones, de hipofunción hipofisaria (1,2). En nuestro estudio, cefaleas, náuseas, vómitos y las alteraciones visuales han sido los síntomas de presentación más frecuentes.
El hipopituitarismo transitorio o permanente es frecuente tras la apoplejía pituitaria y, por ello, un elevado número de pacientes requieren a largo plazo terapia hormonal sustitutiva con esteroides y hormona tiroidea (3,13). En la serie de 37 pacientes descrita por Bills et al (3) la terapia a largo plazo con corticoides u hormona tiroidea fue necesaria en el 89% y el 82% de los casos, respectivamente. Al 11% de los pacientes le fue administrada desmopresina por presentar diabetes insípida. En nuestro grupo, tras una media de seguimiento de 3,5±2,74 años, el 62,5% de los pacientes requirieron terapia sustitutiva con hormona tiroidea y corticoides. En ningún caso se demostró la presencia de hipersecreción hormonal o diabetes insípida.
El TAC Craneal de urgencias demostró la existencia de sangrado en región selar en el 75% (6 de 8) y la presencia de tumoración hipofisaria en el 62,5% (5 de 8) de los casos. En el resto de pacientes el diagnóstico de apoplejía sobre adenoma hipofisario fue realizado en días posteriores al evento, demostrándose la presencia de un macroadenoma en el 100% de los casos (expansión suprasellar en el 55,5% y extensión a seno cavernoso en el 33% de los casos). Los falsos negativos para demostrar la presencia de adenoma y/o sangrado en región selar se debieron a la realización de exploraciones radiológicas no dirigidas específicamente al área selar y a la dificultad de obtener imágenes en planos coronales en pacientes poco colaboradores. Bills and colleagues (3) comprueban, mediante TAC al diagnóstico, la presencia de tumor hipofisario definitivo en el 94% de los pacientes con clínica de AH, mientras que un probable o definitivo infarto o hemorragia sólo se objetiva en el 46% de los casos. Aunque la TAC Craneal puede ser en ocasiones superior a la RMN para visualizar sangrado intratumoral en los primeros días del evento, en la actualidad se considera a la RMN como más sensible tanto en el diagnóstico inicial como en el seguimiento de la hemorragia subaguda (3,6-10).
En nuestra serie, el 100% de los pacientes con afectación de pares craneales recuperaron el normal funcionamiento de éstos durante el seguimiento. McFadzean et al (11) comprueban una mejoría de la paresia ocular en nueve pacientes con apoplejía pituitaria y paresia de pares, alguno de los cuales fueron tratados de forma conservadora. Maccagnagn et al (6) tratan con corticoides (dexametasona 2-15 mg/día) a 7 pacientes con oftalmoplejía por AH, mostrando una progresiva mejoría dentro de una semana de iniciado el tratamiento y recuperación completa de 6 pacientes en 6 semanas. En el trabajo de Bills et al (3) la cirugía es capaz de mejorar la parálisis de pares oculomotores en el 100% de los 30 pacientes afectados, con retorno de la motilidad ocular a la normalidad en el 68%. Los resultados de estos trabajos parecen sugerir que la paresia de pares oculomotores por si sola no constituye un criterio absoluto de cirugía.
Las alteraciones visuales presentes en los dos pacientes no operados de nuestro estudio evolucionaron favorablemente hasta la completa resolución. A su vez, tres de los cuatro pacientes con alteraciones visuales que fueron intervenidos mostraron durante el seguimiento estudio campimétrico y de agudeza visual dentro de la normalidad (en un caso se mantiene una discreta reducción concéntrica del campo visual). McFadzean et al, (11) en un trabajo dirigido a describir los efectos de la apoplejía pituitaria sobre la visión, comunicó que 10 de los 13 pacientes con reducción de agudeza visual mejoraron de tal alteración. Seis de sus pacientes fueron tratados con corticoides y demostraron similar grado de mejora comparados con aquellos sometidos a descompresión quirúrgica, aunque este último grupo presentaba mayor afectación de la agudeza visual al diagnóstico. En la serie de Bills et al (3) la cirugía resultó en una mejora de los déficits de agudeza y del campo visual en un 88% y en un 95%, respectivamente. La mejora en la agudeza visual fue significativamente mayor en aquellos sometidos a cirugía descompresiva durante la primera semana, con una completa resolución en todos los pacientes. No existieron diferencias significativas en la resolución de los déficits del campo o agudeza visual, o de la paresia de pares en aquellos que fueron sometidos a cirugía en los 3 primeros días comparado con el grupo del 4-7 día (3). Maccagnagn et al (6) realizaron un estudio prospectivo para analizar las ventajas y limitaciones del tratamiento médico en la AH, estudiando los casos de 12 pacientes con apoplejía. El protocolo de estudio incluía el tratamiento inicial con dexametasona (2-15 mg/día) durante al menos 1 semana, recurriéndose a la descompresión quirúrgica si en este plazo de tiempo no mejoraba la pérdida de visión o el deterioro del nivel de conciencia. Siete de doce pacientes no requirieron cirugía, demostrándose durante el seguimiento resolución del tumor en el 50%. Los restantes cinco pacientes (dos con ceguera unilateral y tres con ceguera bilateral) requirieron intervención quirúrgica por presentar respuesta desfavorable al trata miento con corticoides.
El manejo óptimo de los episodios agudos de apoplejía pituitaria constituye en la actualidad un foro continuo de discusión entre los grupos con más experiencia en este campo, particularmente por la gran variabilidad del curso clínico de esta condición y por la limitada experiencia individual. Debido a que la presentación clínica de la apoplejía pituitaria comprende un amplio espectro de pacientes que van desde sujetos asintomáticos, cuyo diagnóstico se establece retrospectivamente, a pacientes críticos que presentan pérdida de visión, oftalmoplejía y hemorragia subaracnoidea, es concebible que la mejor forma de tratamiento sea individual y determinada por la severidad de la presentación clínica y la evolución. Los resultados de los estudios más representativos de la literatura parecen sugerir la descompresión quirúrgica precoz (en los primeros 7 días), pero no necesariamente urgente, en aquellos casos que se presentan con importante deterioro visual, disminución del nivel de conciencia o con escasa mejoría tras tratamiento conservador con corticoides. Una excepción sería aquellos pacientes con súbito y severo deterioro visual, en los cuales se recomienda una descompresión quirúrgica urgente (3-5, 14).
AGRADECIMIENTOS
Agradecemos a Francisco Tamayo del Pino su colaboración en la realización y revelado de las fotografías.
BIBLIOGRAFÍA
1. Rolih CA, Ober KP. Pituitary Apoplexy. End Met Clin North Am 1993; 22: 291-302 [ Links ]
2. Thorner MO, Vance ML, Laws ER, Horvath E, Kovacs K. The anterior Pituitary. En: Wilson JD, Foster DW, editores. Williams Textbook of endocrinology, 9th ed. Filadelfia: Saunders 1998; 322-3. [ Links ]
3. Bills DC, Meyer FB, Laws ER, Davis DH, Ebersold MJ. A Retrospective Analysis of Pituitary Apoplexy. Neurosurgery 1993; 33: 602-9. [ Links ]
4. Cardoso ER, Peterson EW. Pituitary apoplexy: A review. Neurosurgery 1984; 14: 363-73. [ Links ]
5. Fernández JM, Villabona CM, Soler J. Apoplejía hipofisaria. Med Clin (Barc) 1991; 97: 589-95. [ Links ]
6. Maccagnan P, Macedo CL, Kayath MJ, Nogueira RG, Abucham J. Conservative management of pituitary apoplexy: a prospective study. J Clin Endocrinol Metab 1995; 80: 2190-7. [ Links ]
7. Fernández JM, Villabona CM, Montaña E, Acebes JJ. Apoplejía hipofisaria: Análisis clínicorradiológico y evolutivo de 18 pacientes. Neurología 1993; 8(1): 4-7. [ Links ]
8. Mohanty S, Tandon PN, Banerji AK, Prahash B. Haemorrhage into pituitary adenomas. J Neurol Neurosurg Psy 1977; 40: 987-91. [ Links ]
9. García-Asencio S, Barrena R, Guelbenzu S, Guedea A, Mota J, Cacicedo Y. Magnetic resonance imaging usefulness in the diagnosis of intratumor bleeding in hypophyseal adenomas. Rev Neurol 1996; 24(134): 1237-40. [ Links ]
10. Lazaro CM, Guo WY, Sami M, Hindmarsch T, Ericson K. Haemorragic pituitary tumours. Neuroradiology 1994; 36(2): 111-4. [ Links ]
11. Mc Fadzean RM, Doyle D, Rampling R, Teasdale E. Pituitary apoplexy and its effect on vision. Neurosurgery 1991; 29: 675. [ Links ]
12. Onesti ST, Wisniewski T, Post KD. Clinical versus subclinical pituitary apoplexy: Presentation, surgical management, and come in 21 patiens. Neurosurgery 1990; 26: 980-6. [ Links ]
13. Pelkonen R, Kuusisto A, Salmi J, Eistola P, Raitta C. Pituitary function after pituitary apoplexy. Am J Med 1978; 65: 773-8. [ Links ]
14. Peter M, De Tribolet N. Visual outcome after transsphenoidal surgery for pituitary adenomas. Br J Neurosurgery 1995; 9(2): 151-7. [ Links ]

