










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.19 no.3 mar. 2002
NOTA CLÍNICA
Erupción cutánea eritematosa como primera manifestación
de leucemia linfática prolinfocítica de células T
L. Beltrán Fernández, B. di Martino Ortiz, J. GiI Herrera, J.A. López- Herce*,
J. de Portugal Alvarez*, J. Menárguez Palanca
Servicios de Anatomía Patológica e Inmunología. *Servicio de Medicina Interna.
Hospital General Universitario Gregorio Marañón. Madrid.
RESUMEN
Se presenta el caso de un varón de 84 años que debutó con una erupción eritematosa y en el cual se efectuó el diagnóstico de leucemia prolinfocítica T. Con este motivo, se revisan diversos aspectos actuales concernientes a la biología, diagnóstico, clasificación pronóstico y tratamiento de las leucemias de células T maduras.
PALABRAS CLAVE: Leucemia linfática prolinfocítica de células T (LPL-T). Leucemia de células T maduras. Lesión cutánea eritematosa. TCL-1.
Cutaneous involvement as the first manifestation of T-cell prolymphocytic leukaemia
ABSTRACT
We present a case of a man 84 years-old, whose presentation feature was a cutaneous inespecific rash, and was diagnosed of T prolymphocytic leukaemia (T-PLL). In this review we analize actual aspects concerning biology, diagnosis, classification, prognosis and treatment of this rare mature T cell leukaemia.
KEY WORDS: T cell chronic prolymphocytic leukaemia (T-PLL). Mature T cell leukaemia. Eritematous cutaneous rash. TCL-1
Trabajo aceptado: 28 de julio de 2000
Correspondencia: L. Beltrán. C/ Estrecho de Mesina nº7, V-2. 28043 Madrid.
INTRODUCCIÓN
La leucemia linfática prolinfocítica de células T (LPL-T) es una neoplasia hematológica infrecuente, de identificación relativamente reciente. Se caracteriza por presentar un curso clínico agresivo con linfocitosis elevada y afectación hepato-esplénica, ganglionar y cutánea, siendo precisamente una erupción cutánea el primer síntoma de nuestro caso. La respuesta a los agentes quimioterápicos es pobre. A continuación presentamos un caso, con cuyo motivo revisamos brevemente diferentes aspectos de la enfermedad.
CASO APORTADO
Varón de 84 años con antecedentes de enfermedad pulmonar obstructiva crónica (EPOC) e hipertensión arterial (HTA) idiopática, portador de virus C de la hepatitis (VHC). Consulta por erupción eritematosa no descamativa en cara, cuello y brazos. A la exploración física destaca la presencia de una masa semisólida en cuello, adenopatías látero-cervicales y axilares y edema de miembros inferiores. En la radiografía de tórax se aprecia derrame pleural izquierdo con atelectasia del lóbulo inferior. En la analítica llama la atención una leucocitosis de 168.000 U/L con linfocitosis absoluta (80%), la hemoglobina es de 15,8 g/L y el recuento plaquetario de 164.000 U/L. El paciente fallece a los 20 días de iniciarse el cuadro, presentando fiebre elevada con severo deterioro de la función pulmonar.
Estudio necrópsico
En la autopsia se encontraron adenopatías generalizadas (mediastínicas, axilares, abdominales, retroperitoneales y pélvicas), así como discreta hepatoesplenomegalia. El estudio histológico de las adenopatías mostró una proliferación neoplásica que crecía en sábana, sin imágenes pseudofoliculares, desplazando los folículos linfoides e infiltrando la cápsula del órgano y la grasa adyacente. Aparecía bien vascularizada, con frecuentes vasos de tipo postcapilar. Citológicamente, se trataba de células discretamente pleomorfas, generalmente de pequeño tamaño, de núcleo redondeado o discretamente indentados, sin nucléolos hipertróficos. El estudio citológico no permitió demostrar la presencia de gránulos azurófilos intracitoplásmicos. El perfil obtenido mediante técnicas inmunohistoquímicas y de citometría de flujo mostró que se trataba de elementos CD3/CD4/CD5/CD45RO/TCRαβ/BCL2+ y negativos para Tdt, cIg, CD20, CD79a, CD10, CD21, CD23, CD1a, CD2, CD8, CD56, CD43, CD30, proteína S100 y P53. La tasa de proliferación celular (Ki67) era al menos de un 50% (Figs. 1 y 2).
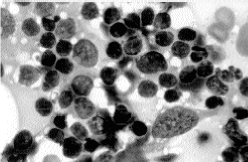
Fig. 1. Las improntas de las adenopatías muestran células discretamente pleomorfas, de pequeño tamaño, de núcleo redondeado sin nucleolo evidente ni granulaciones intracitoplásmicas.

Fig. 2. Corte histológico de una adenopatía latero-cervical en la que mediante técnicas de inmuno histoquímica
para la detección de CD3 se marcan positivamente las células tumorales (fenotipo T), siendo negativos
los centros germinales (fenotipo B).
El parénquima hepático se encontraba extensamente infiltrado a nivel portosinusal. A nivel esplénico, la infiltración es también muy extensa y con un predominio neto en pulpa roja. El parénquima medular aparecía infiltrado de forma difusa. Las áreas eritematosas se correspondían con una infiltración dermo-hipodérmica severa, sin fenómenos de epidermotropismo. En el resto de la economía existían frecuentes imágenes sugerentes de corresponder a leucostasis, sobre todo a nivel pulmonar.
DISCUSIÓN
La leucemia linfática prolinfocítica de células T (LPL-T) y la leucemia de linfocitos grandes granulares (LLG) son dos procesos linfoproliferativos de estirpe T englobados inicialmente dentro de las leucemias linfáticas crónicas de células T (LLC-T). Fue a raíz de la publicación de Brouet y colaboradores (1) en 1975 cuando se comenzó a emplear el término LPL-T para aquellos casos de leucemia de linfocitos T maduros con la morfología inicialmente descrita para LPL (2), reteniéndose el término LLC-T para los casos de LLG. Sin embargo, esta situación originó cierto grado de confusión ya que algunos autores seguían incluyendo todos los casos de neoplasias maduras de estirpe T dentro de la categoría LLC-T, sin distinción entre LLG y LPL-T. Probablemente, la mayoría de casos de LLC-T corresponden realmente a una variante de células pequeñas de LPL-T, ya que presentan las mismas características citogenéticas (alteraciones del 14q32.1) y un curso igualmente agresivo. Por este motivo la LLC-T no aparece recogida en la nueva clasificación de neoplasias linfoides propuesta por la Organización Mundial de la Salud (OMS) (3).
La LPL-T representa aproximadamente un tercio de las leucemias maduras de fenotipo T, que a su vez suponen el 10% de todas las leucemias de fenotipo maduro (4). La LPL es una neoplasia hematológica que se caracteriza por la acumulación de prolinfocitos en sangre periférica e infiltración de médula ósea, bazo, ganglios linfáticos y en ocasiones piel. En las dos terceras partes de los casos las células son de estirpe B (LPL-B). El fenotipo T se asocia a un curso más agresivo (5). Puede surgir de forma esporádica o bien en el seno casos de ataxia-telangiectasia (AT). Clínicamente la LPL-T se caracteriza por esplenomegalia marcada, linfadenopatías y hepatomegalia. La infiltración cutánea se da en un 27% de los casos, normalmente en forma de nódulos aislados, rash máculo-papular o lesiones ampollosas, aunque también se han descrito casos de eritrodermia. La cara y orejas parecen ser localizaciones típicas en la infiltración cutánea por leucemias de células T maduras (6). Histológicamente los infiltrados cutáneos no muestran el epidermotropismo característico del síndrome de Sézary. Se han comunicado casos aislados en los que esta entidad se asocia a poliartritis seronegativa, que es por otra parte un hallazgo frecuente en LLG y en la leucemia/linfoma T del adulto (7). El hallazgo de laboratorio más frecuente es la leucocitosis marcada, con cifras por encima de 100.000 U/L. La anemia y la trombopenia son también relativamente frecuentes (8).
El frotis de sangre periférica es el mejor método diagnóstico en esta entidad. La mayoría de las células son prolinfocitos T en los que se puede apreciar un nucleolo central bien definido. Morfológicamente, son difíciles de diferenciar de los prolinfocitos B; aquéllos tienden a ser más pequeños y a tener un citoplasma más escaso; en la mitad de los casos el contorno nuclear es irregular aunque no es frecuente encontrar núcleos polilobulados o indentaciones profundas. Se ha descrito una variante en la que las células son de menor tamaño, mostrando una cromatina condensada; en estos casos, el nucleolo no es apreciable mediante microscopía óptica. Dentro de esta última variante se puede encuadrar probablemente el caso descrito (4).
La morfología celular no es suficiente para diferenciar el fenotipo (B o T) en esta patología, de ahí la importancia de las técnicas de inmunohistoquímica. En la LPL-T las células son CD4+/CD8- en el 65% de los casos, CD8+/CD4- en el 13% y CD4+/CD8+ en el 21%; además, son CD2, CD7, CD3 y TCR α/β+. La positividad de CD8 en estos últimos casos abre la posibilidad de incluir este fenotipo dentro de un subtipo distinto que se asocia a características clínicas y pronósticas sensiblemente diferentes (9). Como en otros trastornos post-tímicos de células T, las células de la LPL-T son Tdt y CD1a-.
La LPL-T es una neoplasia muy agresiva con una media de supervivencia que varía entre 7 y 13 meses (5,8). Su pronóstico es peor que el de la LPL-B y responde pobremente a la quimioterapia. Se han observado dos formas de presentación: la forma inicialmente progresiva y una forma bifásica, menos frecuente. En este último grupo el curso es inicialmente indolente y tras un periodo variable (media de 33 meses) se produce una fase de transformación haciéndose rápidamente progresiva. Esto sugiere una fase silente preleucémica en la mayoría de los casos esporádicos de LPL-T de forma similar a lo que ocurre con los casos asociados a ataxia-telangiectasia, donde la evolución bifásica es lo más habitual (10). Se ha comunicado un caso de remisión completa de forma espontánea a los 39 meses del diagnóstico que se presentó de forma inicialmente progresiva, con desaparición de la banda de TCRb y normalización de los datos hematológicos (11).
La resistencia a los agentes quimioterápicos es la regla, si bien los análogos de la purina como la fludarabina, deoxicoformicina y clorodeoxiadenosina han supuesto un notable avance en su tratamiento. Aunque los resultados varían considerablemente de un estudio a otro, se han comunicado casos de remisión completa con 2-clorodeoxiadenosina (5). Se han publicado estudios que ofrecen resultados esperanzadores en el tratamiento de la LPL-T con anticuerpo anti-CD52 (CAMPATH-I) (12). El trasplante alogénico de médula ósea supone otra alternativa terapéutica teniendo en cuenta la publicación de Collins y colaboradores (13).
La etiología de esta entidad es desconocida. Recientemente se ha demostrado la presencia de material genético de HTLV-I defectuoso mediante PCR en el DNA extraído de células de sangre periférica y de ganglios linfáticos infiltrados por la neoplasia. Este hallazgo posibilita una nueva línea de investigación aunque el posible papel etiopatogénico de HTLV-I en esta neoplasia es hasta el momento una mera hipótesis (14).
Alteraciones cromosómicas
Como se comentó anteriormente, las LPL-T suelen surgir de forma esporádica o bien el seno de ataxia telangiectasia (AT), enfermedad asociada a mutaciones del gen ATM. Las alteraciones del 14q32.1 suelen consistir en inversiones-duplicaciones centroméricas [inv(14)(q11q32.1/3)] en la AT o traslocaciones simples teloméricas balanceadas [t(14;14) (q11;q32), t(X;14) (q28;q11), t(7;14) (q35;q32)] en casos esporádicos y en ATM15. Traslocaciones similares se han descrito también en casos de proliferaciones T no clonales en AT (16). En casi todos los casos, afectan a una misma región, de unos 160 kb de longitud. En esta región se identificaron originalmente TCL1 y TCL1b (TML1) y más recientemente, TCL6 (17,18). En la región Xq28, se identificó MTPC1, análogo de TCL1 y TCL1b (19).
Los productos génicos correspondientes, TCL1 (p14), TCL1/MTPC1-like (TCL1b, TML1) y MTCP1 (p13) son proteínas protooncogénicas relacionadas (comparten un 40% de su secuencia aa) que se sobreexpresan típicamente en el 90% de las LPL-T como consecuencia de traslocaciones que implican a uno de los genes del TCR (generalmente TCRa/d en 14q11) y a 14q32 (TCL1, TCL1b) y Xq28 (MTCP1) respectivamente. En esta situación se produce sobreexpresión de las proteínas correspondientes al posicionarse bajo la influencia de los enhancers de los TCR correspondientes. Su carácter oncogénico se ha demostrado mediante animales transgénicos, los cuales desarrollan leucemias T maduras tras un periodo de latencia de unos 20 meses (20,21,22).
La función de las proteínas es desconocida. Se conocen datos acerca de la expresión normal de TCL-1, encontrándose expresión de TCL1 fisiológicamente a lo largo de la mayor parte de la evolución de las células B, desde el estadío pre-B hasta el estadio pre-célula plasmática. La expresión más intensa se sitúa a nivel del manto folicular. La mayor parte de los linfomas B pueden expresarla en mayor o menor grado, pero es raro detectarla en linfomas T salvo en el caso de las LPL-T. Su expresión puede ser citoplásmica y nuclear (15,23).
MTCP1 (Xq28) comparte con CDK4a (p16INK4A/p19ARF) el hecho de ser uno los raros locus que codifica más de una proteína, en este caso una proteína mitocondrial (p8MTCP1), cada una con su propio promotor. TCL1, TCL1b (14q32) y MTCP1 (Xq28) se originaron probablemente por duplicación génica (14q32) e inserción (Xq28) en el locus p8 MTCP1 (19).
La resolución de la estructura cristalina de MTCP-1 (24) ha mostrado una arquitectura peculiar, con 8 estructuras en barril constituidas por cadenas β agrupadas de forma com-pacta. Se trata de una estructura probablemente compartida con TCL1, que recuerda a la de la superfamilia lipocaína-calicina, un grupo de proteínas con funciones muy diversas, relacionadas con el transporte de ligandos hidrofóbos de bajo peso molecular (hormonas, PG.) y con la regulación celular a muy diverso nivel.
La identificación en esta misma zona 14q32 del locus TCL6, 7 kb en sentido 5´ del TCL1b (TML1) mediante técnica de RACE, ha permitido identificar al menos 11 isoformas y 5 marcos de lectura abierta generados mediante splicing alternativo, así como en algunas ocasiones, tránscritos de fusión TCL6/TCL1b (17). La función de TCL6 es desconocida. Las ORF analizadas no muestran analogías conocidas con otras secuencias de los bancos. La señalización de los transcritos obtenidos en LPL-T los sitúa a nivel nuclear y/o citoplásmico. Se expresa normalmente en placenta, riñón y en menor grado en amígdala, bazo y ganglio (RT-PCR). En tumores se expresa en linfomas de Burkitt (y líneas Daudi), en algunas LLA-B, pero no en leucemias T sin la t14q32.
En resumen, la LPL-T es una entidad aceptablemente homogénea desde el punto de vista clínico-patológico y molecular, con un mal pronóstico, que debe ser sospechada ante la aparición de linfocitosis y patología cutánea y que requiere una actitud terapéutica enérgica.
Bibliografía
1. Brouet J-C, Flandrin G, Sasportes M, Preud´Homme J-L, Seligmann M. Chronic Lymphocytic Leukemia of T cell origin. Immunological and clinical evaluation in 12 patients. Lancet 1975; 11: 890-3. [ Links ]
2. Galton DAG, Goldman JM, Wiltshaw E, Catovsky D, Henry K, Goldenberg GJ. Prolymphocytic Leukemia. Br J Haematol 1974; 27: 7-23. [ Links ]
3. Lee Harris N, Jaffe ES, et al. World Health Organization Classification of Neoplastic Diseases of the Hematopoietic and Lymphoid Tissues. Jour Clin Onc 1999; 17: 3835-49. [ Links ]
4. Matutes E, Catovsky D. Nature T-cell leukemias and leukemia/lymphoma syndromes. Review of our experience in 175 cases. Leuk Lymph 1991; 4: 81-91. [ Links ]
5. Barrtlet NL, Longo DL. T-Small Lymphocyte Disorders. Semin Hematol 1999; 36: 164-70. [ Links ]
6. Serra A, Estrach MT, et al. Cutaneous Involvement as the First Manifestation in a case of T-Cell Prolymphocytic Leukaemia. Acta Derm Venerol (Stockh) 1998; 78: 198-200. [ Links ]
7. Dybjer A, Hellquist L, Johansson B, Rydgren L, Billstrom R. Seropositive polyarthritis and skin manifestations in T-prolymphocytic leukaemia/Sezary cell leukaemia variant. Leuk Lymphoma 2000; 37 (3-4): 437-40. [ Links ]
8. Hoyer JD, Ross CW, Li CY, Witzig TE, Gascoyne RD, Dewald GW, et al. Hanson. True T-Cell Chronic Lymphocytic Leukemia: A Morphologic and Immunophenotypic Study of 25 Cases. Blood, 1995; 86: 1163-69. [ Links ]
9. Ascani S, Leoni P, Fraternali Orcioni G, Bearzi I, Picciolini M, Materazzi M, et al. T-cell prolymphocytic leukemia: does the expression of CD8+ phenotype justify the identification of a new subtype? Description of two cases and review of the literature. Ann Oncol 1999; 10: 649-53. [ Links ]
10. Garand R, Goasguen J, Brizard A, Buisine J, Charpentier A, Claisse JF, et al. Indolent course as a relatively frequent presentation in T-prolymphocytic leukemia. Br J Haematol 1998; 103, 488-94. [ Links ]
11. Shichishima T, Kawaguchi M. T-prolymphocytic leukemia with spontaneous remission. Br J Haematol 2000; 108: 397-9. [ Links ]
12. Pawson R, Dyer MJS, Barge R, et al. Treatment of T-cell prolymphocytic leukemia with human CD52 antibody. J Clin Oncol 1997; 15: 2667-72. [ Links ]
13. Collins RH, Piñeiro LA, Augura ED, et al: Treatment of T-prolymphocytic leukemia with allogenic bone marrow transplantation. Bone Marrow Transplant 1998; 21: 627-8. [ Links ]
14. Kojiyama K, Hara M, Sawada T, Miyata A, Saito H, Matsuo Y, et al. Human T-lymphotropic virus type I provirus and T-cell prolymphocytic leukemia. Leuk Lymphoma 2000; 38: 381-6. [ Links ]
15. Thick J, Metcalfe JA, Mak Y-F, Beatty D, Minegishi M, Dyer MJS, et al. Expression of either the TCL1 oncogene, or transcripts from its homologue MTCP/c6.1B, in leukaemic and non-leukaemic T cells from ataxia telangiectasia patients. Oncogene 1996; 12: 379-86. [ Links ]
16. Britto-Babapulle V, Catovsky D. Inversion and tandem duplication involving chromosome 14q11 and 14q32 in T- prolymphocytic leukaemia in patients with ataxia- teleangiectasia. Cancer Genet Cytogenet 1991; 55: 1-9. [ Links ]
17. Virgilio L, Narducci MG, Isobe M, Billips LG, Cooper MD, Croce CM, et al. Identification of the TCL1 gene involved in T- cell malignancies. Pro Natl Acad Sci USA 1994; 91: 12530-4. [ Links ]
18. Saitu M, Sugimoto J, Hatakeyama T, Russo G, Isobe M. Identification of the TCL6 genes within the breakpoint cluster region on chromosome 14q32 in T-cell leukemia. Oncogene 2000; 19: 2796-802. [ Links ]
19. Gritti C, Choukron V, Soulier J, Madani A, Dastot H, Leblond V, et al. Alternative origin of p13MTCP1 -encoding transcripts in mature T-cell proliferations with t(X;14) translocations. Oncogene 1997; 15: 1329-35. [ Links ]
20. Virgilio L, Lazzeri C, Bichi R, Nibu K, Narducci M, Russi G, et al. Deregulated expression of TCL1 causes T-cell leukemia in mice. Proc Natl Acad Sci USA 1998; 95: 3885-9. [ Links ]
21. Gritti C, Dasdot H, Soulier J, Janin A, Daniel MT, Madani A, et al. Transgenic mice for MTCP1 develop T- cell prolymphocytic leukemia. Blood 1998; 92: 368-73. [ Links ]
22. Chiali, Gritti C. Transgenic mice expressing MTCP1: an animal model for T-cell prolymphocytic leukemia. Ann Pharm Fr 2000; 58 (1): 54-61. [ Links ]
23. Narducci MG, Pescarmona E, Lazzeri C, Signoretti S, Lavinia AM, Remotti D, et al. Regulation of TCL1 Expression in B- and T-Cell Lymphomas and Reactive Lymphoid Tissues. Cancer Res 2000; 60: 2095-100. [ Links ]
24. Fu ZQ, Du Bois GC, Song SP, Kilokovskaya I, Virgilio L, Rothstein JL, et al. Crystal structure of MTCP-1: implications for role of TCL-1 and MTCP-1 in T cell malignancies. Proc Natl Acad Sci USA 1998; 95: 3413-8.

