










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkAnales de Medicina Interna
Print version ISSN 0212-7199
An. Med. Interna (Madrid) vol.19 n.5 May. 2002
CARTAS AL DIRECTOR
Dolor torácico y marcada elevación enzimática.
Forma de presentación de la enfermedad de Mc Ardle
Sr. Director:
Hemos leído con interés el artículo publicado en su revista de López Martin y cols. (l) "Enfermedad de Mc Ardle: descripción de cuatro hermanos con defícit de miofosforilasa". Hemos tenido la oportunidad de diagnosticar un nuevo caso en una paciente de edad avanzada, que acudió a nuestro hospital por intenso dolor torácico, objetivándose cifras muy elevadas de CPK por lo que, en un principio, se pensó en la posibilidad de patología isquémica cardíaca, y que a continuación se comenta:
Se trataba de una mujer de 64 años que ingresó por un cuadro de dolor torácico opresivo, irradiado a hombros, que no tenía relación con el ejercicio ni se modi;caba con la postura y que se repitió, en varias ocasiones, sin acompañarse de cortejo vegetativo. También, refería debilidad muscular generalizada de forma episódica, pero principanente en relación con el ejercicio. Entre sus antecedentes destacaba el hecho de ser consultada en dos ocasiones, hacía 12 y 2 años, por diversos motivos apreciándose alteraciones de enzimas musculares, a las que no se les dio mayor importancia, sin que tampoco se llegara a estudiar en ese sentido. En la exploración física, presentaba fuerza muscular disminuída globalmente, sobre todo a nivel de la cintura pelviana y, en menor medida, en la escapular. En los datos complementarios: hemograma normal. VSG de 30 mm a la primera hora. Bioquímica: GOT 630 UVL (N: 1-37), GPT 470 UVL (N: 1-40), GGT 147 UVL (N: 1-39), CPK 4785 UVL (N: 0-170), CK-MB 12 UVL (N: 0-24), el resto de los parámetros bioquimicos, los de coagulación y las hormonas tiroideas fueron normales. Las serologías de hepatitis B, C y D fueron negativas. En diversos electrocardiogramas presentaba bradicardia sinusal. Se realizó una electromiografía en la que los hallazgos obtenidos fueron sugestivos de un proceso tipo miopatía inflamatoria. El estudio histopatológico, incluyendo examen ultraestructural (Fig. l) de biopsia del músculo deltoides izquierdo, fue indicativo de enfermedad por depósito de glucógeno al evidenciarse cambios compatibles con miopatía glucogenósica, con ausencia de miofosforilasa, compatible con con glucogenosis tipo V- Enfermedad de Mc Ardle. Realizadas las determinaciones de mioglobina en sangre y orina, de piruvato y lactato (antes y despues del ejercicio) y la prueba de sobrecarga oral de glucosa, pudimos constatar la presencia de mioglobina en orina -sin datos de insuficiencia renal ausencia de subida de lactato en sangre tras el ejercicio y una mayor tolerancia al ejercicio tras la administración oral de glucosa.
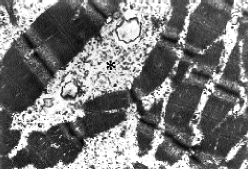
Fig. 1. Imagen ultraestructural que muestra sustitución de las miofibrillas
por gránulos de glucógeno (asterisco) (6000 x).
La Enfermedad de McArdle (glucogenosis tipo V) es una enfermedad poco frecuente que se origina por un déficit de miofosforilasa (2,3). Generalmente, la enfermedad se manifiesta antes de los 30 años (4,5), aunque también se han descrito en la infancia y de aparición tardía (6,7) como es nuestro caso, en el que destaca tanto la forma de presentación en lo que se refiere a la edad (aparición tardía), como a la clínica que hizo pensar, inicialmente, en infarto agudo de miocardio. No obstante, rehistoriando a la paciente, se encuentran datos clínicos y analíticos previos que hubieran podido hacer el diagnóstico años antes si se hubiese pensado en esta enfermedad.
Es conocido que esta forma de aparición tardía puede presentar alguna peculiaridad clínica como marcada debilidad muscular, ausencia de calambres, etc. (7,8). Así se detecta en el caso descrito en el que, la ausencia de anormalidades electrocardiográficas en registros seriados, la escasa elevación de la CK-MB y la propia evolución de la paciente, descartaron patología isquémica coronaria y se planteó el estudio de una paciente con miopatía, siendo el resultado de la electromiografía de miopatía inflamatoria. Se barajaron otras posibilidades diagnósticas como otras miopatías metabólicas o la polimiositis (entidades con las que se confunde ocasionalmente) y con las que hay que establecer el diagnóstico diferencial (9) siendo definitivo, en cuanto al diagnóstico, la biopsia muscular en la que se objetivó la ausencia total de miofosforilasa.
Para concluir, en pacientes de edad avanzada con debilidad muscular y enzimas musculares elevadas, pese a su carácter excepcional de presentación, debe tenerse en cuenta la enfermedad de McArdle en el diagnóstico diferencial.
I. Rodríguez López, F. L. Lado Lado, E. Donado Budiño, S. Lojo Rocamonde*, E. Pintos Martínez**
Servicios de Medicina Interna. *Laboratorio y **Anatomía Patológica. Complejo Hospitalario Universitario de Santiago. Departamento de Medicina. Universidad de Santiago de Compostela.
1. López Martin A, Baños Madrid RI, Garcia-Estañ Candela J, García Pérez B, Pérez Bautista FJ, Salmerón P. Enfermedad de Mc Ardle: descripción de cuatro hermanos con déficit de miofosforilasa. An Med Interna (Madrid) 2001; 18: 136-8.
2. Mc Ardle B. Myopathy due to a defect in muscle glycogen breakdown. Clin Sci 1951; 10: 13-33.
3. Schmidt R, Mahler R. Chronic progressive myopathy with myoglobinuria. Demonstration of a glycogenolytic defect in the muscle. J Clin Invest 1959; 38: 2044-58.
4. Cornelio F, Di Donato S. Myopathies due to enzyme def1ciencies. J Neurol 1985; 232: 329-40.
5. DiMauro S, Bresolin N. Phosphorylase deficiency. En: Engel AG, Banker BQ, eds. Myology. Nueva York: McGraw-Hill, 1986:1585-1601.
6. Williams J, Hosking G. Type V glycogen storage disease. Arch Dis Child 1985; 60: 1184-6.
7. Engel WK, Eyerman EL, Williams HE. Late-onset type of skeletal-muscle phosphorylase def1ciency. A new familial variety with completely and partially affected subjetcs. N Engl J Med 1963; 260: 135-7.
8. Felice KJ, Schneebaum AB, Jones HR Jr. Mc Ardle's disease with late-onset symptoms: case report and review of the literature. J Neurol Neurosurg Psychiatry 1992;55:407-408.
9. Formigo E, Gómez N, Ferreiro JL, Anton L. Retraso en el diagnóstico de la enfermedad de McArdle. An Med Interna (Madrid) 1994; 11: 136-8.

