










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.19 no.7 jul. 2002
NOTA CLÍNICA
Fallo hepático fulminante asociado al uso de cocaína
J. Campos Franco, C. Martínez Rey, E. Pérez Becerra*, A. González Quintela
Servicio de Medicina Interna. Servicio de Anatomía Patológica*. Hospital Clínico.
Complexo Hospitalario Universitario. Santiago de Compostela
RESUMEN
Una paciente de 23 años presentó datos bioquímicos de hepatitis aguda grave asociados a confusión y asterixis pocas horas después de esnifar una dosis elevada de cocaína. Los niveles sanguíneos de cocaína se encontraban muy elevados y en la biopsia hepática realizada pocos dias más tarde se observó necrosis centrolobulillar. La recuperación fue completa con tratamiento conservador. La toxicidad de la cocaína se debe tener en cuenta en casos similares de insuficiencia hepática aguda grave.
PALABRAS CLAVE: Cocaína. Hepatitis. Hepatitis fulminante.
Cocaine-related fulminant liver failure
ABSTRACT
A 23-year-old woman developed biochemical signs of acute severe hepatitis together with confusion and flapping tremor after snorting a large dose of cocaine. Blood levels of cocaine were very high and liver biopsy performed few days later showed centrilobular necrosis. She recovered completely with conservative measures. Cocaine toxicity should be considered in similar cases of fulminant liver failure.
KEY WORDS: Cocaine. Hepatitis. Fulminant hepatitis.
Trabajo aceptado: 04 de enero de 2001
Correspondencia: Joaquín Campos Franco. Servicio de Medicina Interna. Hospital Clínico Universitario. 15706
Santiago de Compostela. Fax: 981-950 501.
INTRODUCCIÓN
La cocaína se extrae de la hoja de la planta Erythroxylon coca y sus propiedades estimulantes y anestésicas ya eran apreciadas por los incas de la América pre-colombina. A finales del siglo XIX Freud describió sus efectos sobre el SNC y posteriormente comenzó a utilizarse ampliamente como anestésico local (1). La cocaína es la principal droga de abuso en la actualidad. El abuso extendido de la cocaína se ha acompañado de un aumento progresivo de las comunicaciones sobre sus efectos adversos, sobre todo alteraciones cardiovasculares (infarto de miocardio, arritmias, rotura de aneurismas), del SNC (accidentes cerebrovasculares, convulsiones, psicosis, alucinaciones), musculares (rabdomiolisis) y otras (infarto intestinal, hipertermia maligna, edema agudo pulmonar, o coagulación intravascular diseminada) (2).
Los fármacos y tóxicos son causas bien conocidas de fallo hepático fulminante. La hepatotoxicidad inducida por cocaína está bien descrita en animales (sobre todo en roedores) (3-6) pero existen muy pocos casos bien documentados de hepatitis aguda grave inducida por cocaína en humanos (7-10). Se presenta un caso de hepatitis fulminante asociada al uso de cocaína.
CASO APORTADO
Una mujer de 23 años fue traida a nuestro Servicio de Urgencias por disminución del nivel de conciencia. A su ingreso la paciente estaba somnolienta y confusa. La temperatura axilar era 36 ºC, la tensión arterial 90/50 mmHg y la frecuencia cardíaca 89 lpm. Presentaba asterixis. No presentaba focalidad neurológica ni signos meníngeos. El resto del examen físico era normal. En sangre periférica el recuento de leucocitos era 12.450/mm3 (90% neutrófilos, 4% linfocitos y 6% monocitos), hemoglobina 11,5 g/dL y plaquetas 301.000/mm3. La AST sérica era 1.092 U/L, ALT 1.013 U/L, GGT 15 U/L, fosfatasas alcalinas 103 U/L, bilirrubina total 1,2 mg/dL, urea 44 mg/dL, creatinina 1,5 mg/dL, sodio 136 mEq/L y potasio 6,1 mEq/L. El índice de Quick era 45%, el TTPA 29,9 seg, el fibrinógeno sérico 334 mg/dL y el factor V 54%. La pO2 arterial era 76 mmHg, pCO2 47 mmHg y pH 7,36. Las serologías del virus de la hepatitis A (IgM), hepatitis B (HBsAg y anti-HBc), hepatitis C, hepatitis E, virus Herpes simplex (IgM), virus Epstein-Barr (IgM) y Citomegalovirus (IgM) fueron negativas. El RNA del virus de la hepatitis C era negativo en suero por PCR. Los anticuerpos antinucleares, antimúsculo liso y antimitocondriales fueron negativos. La cuantificación en suero de IgG, IgA, IgM, hormonas tiroideas, ferritina, ceruloplasmina y alfa-1-antitripsina estaba dentro de la normalidad. La radiografía de tórax, ecografía abdominal y TAC cerebral no revelaron anormalidades. Al ingreso se administró flumazenilo y naloxona intravenosos sin que se produjese cambio alguno en el nivel de conciencia. Se administró paromomicina oral, lactulosa y vitamina K intravenosa. El nivel de conciencia volvió a la normalidad a las 24 horas del ingreso y la asterixis desapareció. La evolución de la ALT sérica y del índice de Quick se muestra en la figura 1. Los niveles de bilirrubina sérica se mantuvieron dentro de la normalidad durante todo el episodio. El análisis toxicológico de una muestra de sangre obtenida al ingreso demostró elevados niveles de cocaína y monoacetil-morfina (19,8 mcg/mL y 236 mcg/mL, respectivamente), trazas de paracetamol y codeína y no se detectaron niveles de alcohol ni benzodiacepinas. Tras recuperarse, la paciente admitió ser consumidora ocasional de drogas. Doce horas antes del ingreso había "esnifado" 1 g de cocaína y 0,25 g de heroína. El mismo día había tomado un comprimido de paracetamol con codeína por dismenorrea. Regularmente tomaba fluracepam por insomnio.
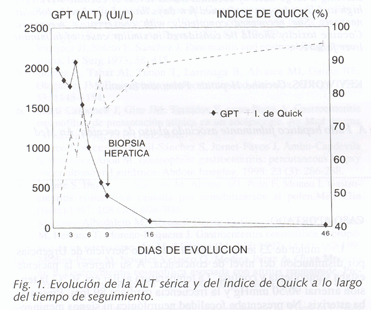
La biopsia hepática percutánea practicada al noveno día de ingreso mostró necrosis centrolobulillar (Fig. 2). La paciente fue dada de alta 12 días después del ingreso. El posterior seguimiento confirmó la total resolución de las anomalías bioquímicas (Fig. 1).

DISCUSIÓN
La toxicidad hepática por cocaína en animales fue descrita ya en 1890 por Ehrlich (3). Aprovechando el interés por esta droga en los años 70, se han realizado múltiples estudios, fundamentalmente en roedores, que han demostrado la capacidad hepatotóxica dosis y tiempo dependiente de la cocaína a través de mecanismos relacionados con el sistema P450. Tras administrar una dosis de cocaína más del 90% es metabolizada por las pseudocolinesterasas plasmáticas (80%) y esterasas hepáticas (10%). Sólo un 10% es metabolizada a través del sistema P450. Sin embargo esta vía metabólica minoritaria produce metabolitos intermedios como la norcocaína, nitróxido de norcocaína , peróxido de hidrógeno y anión superóxido que causan una marcada disminución del NADPH hepatocitario y del glutation. La deplección de glutation y NADPH agota los sistemas antioxidantes celulares; de esta forma peróxido de hidrógeno, anión superóxido y radicales hidroxilo quedan libres para estimular la peroxidación lipídica y dañar las membranas celulares. La peroxidación lipídica y la formación de radicales libres serían la causa de la hepatotoxicidad por cocaína (1,12). En la mayoría de los trabajos en roedores, tras la administración de cocaína se detecta una elevación de las transaminasas y se produce necrosis hepática a nivel periportal (zona 1) (4,12), si bien existen modificaciones dependiendo de la dosis de cocaína, sexo y cepa de los ratones y del uso de agentes inductores o inhibidores del sistema P450 (5). En ratones inducidos previamente con etanol el daño hepático es perivenular (8,13) y en ratones inducidos con fenobarbital la lesión es periportal (5). Las hembras de la cepa B6AF1 presentaron mayor elevación de GOT y mayor necrosis periportal que los machos, ambos inducidos con fenobarbital, tras administrar cocaína (5), corrigiéndose esta mayor susceptibilidad de las hembras tras administrar testosterona. En ratones a los que se administró alcohol y cocaína las lesiones histológicas hepáticas fueron más severas y aumentaron los indicadores de peroxidación lipídica (13-15).
A pesar de esta amplia evidencia de toxicidad en animales existen pocos casos descritos de toxicidad hepática en humanos. En el trabajo original de Marks y Chapple (16) en 1967 sobre 89 consumidores de cocaína y heroína se evidenciaron alteraciones hepáticas en el 80% que remitían tras hospitalización y reaparecían tras el alta relacionándose la mejoría enzimática con la suspensión del consumo durante el ingreso. Resulta llamativo que en trabajos similares recientes (17-19) estos resultados no se reproducen. Probablemente la carencia de marcadores virales en el año 1967 enmascaró a muchos portadores de infecciones crónicas por VHC y VHB. Tras revisar la literatura se encuentra un total de 22 casos de disfunción hepática severa relacionada con la cocaína, de los cuales sólo se dispone de histología hepática en 11 casos, (siete sobre necropsia) (7-10). La mayoría de estos pacientes eran menores de 35 años, y fueron llevados a Urgencias por alteraciones del comportamiento o del nivel de conciencia, detectándose elevación de enzimas hepáticas, alargamiento del tiempo de protrombina, datos de rabdomiolisis y grados variables de insuficiencia renal. La mayoría presenta una rápida normalización de las ALT/AST y de la protrombina a los pocos días; los restantes casos presentan una evolución tórpida con desarrollo de insuficiencia renal, rabdomiolisis y CID, falleciendo en poco tiempo. En todos ellos se detectó la presencia de cocaína en análisis toxicológico de orina, pero en ninguno se cuantificaron los niveles plasmáticos de cocaína. Asimismo, no se descartaron exhaustivamente otras posibles causas de fallo hepático fulminante (7-9). La escasez relativa de casos de hepatotoxicidad grave por cocaína a pesar de su uso extendido sugiere algún modo de susceptibilidad o la asociación de cofactores en los casos graves. Entre estos factores se han sugerido: inducción enzimática previa (por alcohol, fenobarbital, paracetamol) (7,11,17), déficit enzimáticos (pseudocolinesterasa y glucosa 6-P-deshidrogenasa) (1,7,10,11), politoxicomanía (11), asociación de cocaína y opioides (como en nuestro caso) (6), malnutrición (17), o infecciones crónicas víricas (10,11). Respecto al incremento de la hepatotoxicidad de la cocaína por el alcohol si bien en experimentación animal parece claramente probada, los estudios sobre poblaciones humanas no demuestran este efecto sinérgico (17,19). El caso presentado había ingerido concomitantemente paracetamol, pero en dosis que no parecen suficientes para contribuir a la hepatitis aguda grave de la paciente.
De los 11 casos biopsiados de hepatitis aguda grave asociada al uso de cocaína, en seis se encontró necrosis centrolobulillar (zona 3) y midzonal, con esteatosis microvesicular en zona 1 (8-10); en 3 casos se encontró necrosis panlobulillar con lisis masiva de los hepatocitos(8-10); en un caso se evidenció inflamación y necrosis periportal (zona 1) (7); y finalmente en un caso no se encontraron signos de necrosis ni de inflamación relacionándose este silencio histológico con la tardanza en la obtención de la muestra (biopsia obtenida al 8º día de ingreso, frente a las restantes que se obtuvieron a los 3-4 días). Las lesiones hepáticas por cocaína en humanos suelen corresponder por tanto a necrosis centrolobulillar (zona 3, como en el caso presentado), de modo similar a otros tóxicos, como paracetamol. Esta desigual localización de las lesiones en humanos y roedores se debería a diferencias en la distribución lobulillar del sistema enzimático P450 (12). Algunos autores cuestionan la capacidad de la cocaína para producir daño hepático, proponiendo que las alteraciones hepáticas que presentan estos pacientes podrían ser debidas a hipoxia, shock, hipertermia o CID (presentes en alguno de los casos, aunque no en todos, incluido el aquí presentado), circunstancias que, como es bien sabido, pueden producir una imagen histológica similar.
Es frecuente, como ya se ha mencionado, que al daño hepático por cocaína se asocie rabdomiolisis (10,20,21), lo que ha llevado a buscar un mecanismo de lesión común. La cocaína inhibe la recaptación de catecolaminas lo que justifica sus efectos simpaticomiméticos. El potente efecto vasoconstrictor que genera provocaría una disminución del flujo sanguíneo hepático (22) y una isquemia mantenida sobre el músculo esquelético (20) responsable de la rabdomiolisis. Asimismo se ha demostrado que las catecolaminas disminuyen el glutation hepático por mecanismos independientes del sistema P450 (22) y que la cocaína es un tóxico directo para el músculo del ratón in vitro (20).
Se presenta, en resumen, un caso de hepatitis aguda grave con necrosis centrolobulillar demostrada histológicamente asociada al uso de cocaína en el que se descartan otras posibles causas infecciosas, tóxicas, inmunológicas y metabólicas. No se asoció hipertermia, hipoxia ni shock. El descenso del índice de Quick y los datos de encefalopatía hepática permitirían clasificarlo como fallo hepático fulminante de acuerdo con los criterios de O'Grady (23). Es, en nuestro conocimiento, el primer caso en el que se han cuantificado simultáneamente los niveles de cocaína en plasma, que se encontraban marcadamente elevados. Dada la prevalencia de su consumo, pensamos que debe tenerse en cuenta la cocaína como posible agente etiológico en casos similares de hepatitis aguda grave.
Bibliografía
1. Kloss MW, Rosen GM, Rauckman EJ. Cocaine-mediated hepatotoxicity. A critical review. Biochem Pharmacol 1984; 33: 169-73. [ Links ]
2. Cregler LL, Mark H. Medical complications of cocaine abuse. N Engl J Med 1986; 315: 1495-500. [ Links ]
3. Ehrlich P. Studien in der cocainreihe. Deutsch Med Wochenschr 1890; 16: 717-9. [ Links ]
4. Freeman RW, Harbison RD. Hepatic periportal necrosis induced by chronic administration of cocaine. Biochem Pharmacol 1981; 30: 777-83. [ Links ]
5. Thompson ML, Shuster L, Casey E, Kanel GC. Sex and strain differences in response to cocaine. Biochem Pharmacol 1984; 33: 1299-307. [ Links ]
6. Shuster L, Garhart CA, Powers J, Grunfeld Y, Kanel G. Hepatotoxicity of cocaine. En: Clouet D, Asghar K, Brown R, eds. Mechanisms of cocaine abuse and toxicity. Rockville, MD: National Institute of Drug Abuse, 1988: 250-75. [ Links ]
7. Perino LE, Warren GH, Levine JS. Cocaine-induced hepatotoxicity in humans. Gastroenterology 1987; 93: 176-80. [ Links ]
8. Kanel GC, Cassidy W, Shuster L, Reynolds TB. Cocaine-induced licer cell injury : comparison of morphological features in man and in experimental models. Hepatology 1990; 11: 646-51. [ Links ]
9. Wanless IR, Dore S, Gopinath N, et al. Histopathology of cocaine hepatotoxicity. Gastroenterology 1990; 98: 497-501. [ Links ]
10. Silva MO, Roth D, Reddy KR, Fernandez JA, Albores-Saavedra J, Schiff ER. Hepatic dysfunction accompanying acute cocaine intoxication. J Hepatol 1991; 12: 312-5. [ Links ]
11. Mallat A, Dhumeaux D. Cocaine and the liver. J Hepatol 1991; 12: 275-8. [ Links ]
12. Van Thiel D, Perper J. Hepatotoxicity associated with cocaine abuse. En: Marc Galanter ed. Recent Developments in Alcoholism, Volume 10: Alcohol and Cocaine: similarities and differences. Plenum Press. New York. 1992; 335-41. [ Links ]
13. Boyer CS, Petersen D. Potentiation of cocaine-mediated hepatotoxicity by acute and chronic ethanol. Alcohol Clin Exp Res 1990; 14: 28-31. [ Links ]
14. Pirozkhov S, Eskelson C, Watson R. Chronic ethanol and cocaine-induced hepatotoxicity : effects of vitamin E suplementation. Alcohol Clin Exp Res 1992; 16: 904-9. [ Links ]
15. Odeleye OE, Watson RR, Eskelson CD, Earnest D. Enhancement of cocaine-induced hepatotoxicity by ethanol. Drug Alcohol Depend 1993; 31: 253-63. [ Links ]
16. Marks V, Chapple PAL. Hepatic dysfunction in heroin and cocaine users. Br J Addict 1967; 62: 189-95. [ Links ]
17. Tabasco-Minguillan J, Novick DM, Kreek MJ. Liver function tests in non-parenteral cocaine users. Drug Alcohol Depend 1990; 26: 169-74. [ Links ]
18. Kothur R, Marsh F, Posner G. Liver function tests in nonparenteral cocaine users. Arch Intern Med 1991; 151: 1126-8. [ Links ]
19. Worner TM. Hepatotoxicity is not increased in alcoholics with positive urinary cocaine metabolites. Drug Alcohol Depend 1994; 35: 191-5. [ Links ]
20. Herzlich BC, Arsura EL, Pagali M, Grob D. Rhabdomyolysis related to cocaine abuse. Ann Intern Med 1988; 109: 335-6. [ Links ]
21. Roth D, Alarcon FJ, Fernandez JA, Preston RA, Bourgoignie JJ. Acute rhabdomyolysis associated with cocaine intoxication. N Engl J Med 1988; 319: 673-7. [ Links ]
22. James RC, Schiefer MA, Roberts SM, Harbison RD. Antagonism of cocaine-induced hepatotoxicity by the a-adrenergic antagonists phentolamine and yohimbine. J Pharmacol Exp Ther 1987; 242: 726-32. [ Links ]
23. OGrady J, Schalm R, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 33: 273-5. [ Links ]

