










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.19 no.8 ago. 2002
NOTA CLÍNICA
Miopatía por déficit de carnitina: un caso de diagnóstico tardío
M. A. Rico Corral, J. M. de la Vega Vázquez, C. Holgado Silva, A. Aznar Martín, E. Zamora Madaría
Servicio de Medicina Interna C. Hospital Virgen Macarena. Sevilla
RESUMEN
Las miopatías causadas por trastornos del metabolismo lipídico son un grupo patológico poco frecuente, entre las que destacan las asociadas a déficit de carnitina. En este grupo se distinguen dos formas clínicas: la forma miopática, que suele cursar con niveles séricos normales de carnitina y concentraciones descendidas en músculo, y la forma sistémica, que cursa con crisis de encefalopatía y disfunción hepática además de la clínica miopática, y en la que suelen estar descendidas las concentraciones de carnitina en suero, músculo e hígado. Ambas formas muestran en la biopsia acúmulos lipídicos característicos. Presentamos el caso de una forma miopática de déficit de carnitina, excepcional por la edad de la paciente en el momento del diagnóstico, cuya evolución fue desfavorable. Se ha descrito que ciertas modificaciones dietéticas, como el uso de triglicéridos de cadena media, la restricción de grasas o el incremento de carbohidratos de la dieta, así como la prednisona y los suplementos orales de carnitina, pueden producir mejorías clínicas, sin modificar significativamente las concentraciones musculares de carnitina.
PALABRAS CLAVE: Miopatía lipídica. Carnitina. Carnitina-palmitoil-transferasa.
Carnitine deficiency myopathy: a case of late diagnosis
ABSTRACT
Myopathies caused by lipidic metabolism alterations are very infrecuent. Carnitine deficiency-associated myopathies are included in this group. Two main types of carnitine deficiency syndromes have been delineated: a predominantly myopathic form, with normal serum and low muscle carnitine levels, and a systemic form, with encephalopathy, hepatic dysfunction, muscle weakness and low muscle, liver and serum carnitine levels. Both types have typical lipid stores in muscle biopsy. We describe the case of a myopathic form of carnitine deficiency. Due to the age of the patient, this is an unusual case, with an unfavourable evolution. Therapeutic measures used in these patients have included prednisone, carnitine replacement and a low-fat with medium chain tryglycerides and high-carbohydrate diet. However, in none of the patients responding to therapy, a significative increase in muscle carnitine has been demonstrated.
KEY WORDS: Lipidic myopathy. Carnitine. Carnitine-palmityl-transferase.
Trabajo aceptado: 25 de enero de 2001
Correspondencia: Miguel Angel Rico Corral. C/ Milano Govinda, nº 11. 41020 Sevilla. e-mail: marc0001@saludalia.com
INTRODUCCIÓN
El conjunto de enfermedades agrupadas bajo la denominación de miopatías lipídicas constituye un grupo patológico infrecuente, cuyo diagnóstico suele realizarse en las edades infantil o juvenil y para el cual se requiere un estudio ultraestructural e histoquímico de la pieza de biopsia muscular. Presentamos el caso de una miopatía por déficit de carnitina con afectación exclusivamente muscular y no sistémica, excepcional por la edad de la paciente en el momento del diagnóstico.
CASO APORTADO
Mujer de 39 años, que en el interrogatorio refería haber comenzado a sufrir, desde los 12 años de edad, frecuentes caídas al suelo sin trastorno del nivel de conciencia, con dificultad para la incorporación. La sensación de debilidad de miembros inferiores fue haciéndose progresivamente más intensa, localizándose predominantemente en región proximal. A los 28 años comenzó a presentar movimientos oculares incoordinados. En ningún momento solicitó consejo médico por esta sintomatología. Un año antes del ingreso hospitalario en nuestro centro comenzó a apreciar pérdida de la potencia de la voz. Dos meses antes del ingreso presentó un cuadro catarral que evolucionó tórpidamente a pesar de realizar diversos tratamientos antibióticos en régimen ambulatorio. Este cuadro fue progresando hasta llegar a provocar insuficiencia respiratoria grave, que precisó ingreso en la unidad de cuidados intensivos y ventilación mecánica. Fue diagnosticada de neumonía basal derecha. La evolución en la unidad de cuidados intensivos fue tórpida, presentando como complicaciones una sepsis por Pseudomona Aeruginosa y un neumotórax izquierdo yatrógeno. Cuando se consiguió la desconexión de la paciente del respirador y el control del proceso séptico, fue trasladada a nuestro servicio de medicina interna. A la exploración se trataba de una paciente con nivel normal de conciencia, con signos de atrofia muscular en miembros, que presentaba una fuerza de 1/5 a nivel proximal y 3/5 a nivel distal en miembros superiores, y de 2/5 a nivel proximal y 3/5 a nivel distal en los inferiores. Los reflejos miotáticos estaban abolidos y no había signos de piramidalismo. La sensibilidad estaba por completo conservada. Aparecía una piel descamativa y una facies de batracio, con rasgos borrados. Existía una tendencia a la desviación interna del ojo derecho, con discreta quemosis y nistagmus atáxico con la mirada hacia adentro. El murmullo vesicular era normal salvo la apreciación de algunos crepitantes aislados y existía una cánula de traqueostomía permanente. Los tonos cardíacos eran rítmicos y no se apreciaban soplos. No había hallazgos a la palpación abdominal. Por último, existía un pie equino varo bilateral.
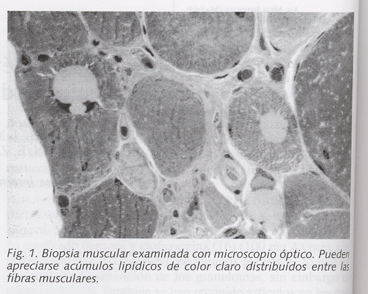
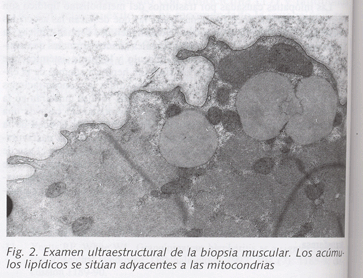
En cuanto a los estudios complementarios, destacaron los hallazgos siguientes: la bioquímica sanguínea, incluyendo medición de colesterol total y sus fracciones así como triglicéridos, fue repetidamente normal, salvo incrementos intermitentes de CPK total (valores entre 230 y 380 U/l, con porcentaje de fracción MB inferior al 10% del total) con aldolasa normal, mínimos incrementos de LDH y en una ocasión un valor de ALT de 70 U/l. La bioquímica urinaria fue igualmente normal, incluyendo la determinación de pH, y apareciendo una discreta leucocituria en el sedimento mientras la paciente permanecía con sondaje vesical. Las determinaciónes de cuerpos cetónicos y mioglobinuria fueron repetidamente negativas. La serología para virus de la hepatitis A, B y C fue negativa. El estudio hematológico mostraba un fibrinógeno de 642 mg/dl y una VSG de 45 mm, con recuentos normales en las tres series celulares. La determinación de hormonas tiroideas mostró valores normales. Las oximetrías mostraban moderadas hipoxemias con valores normales de bicarbonato y pH. En el electrocardiograma se apreciaba ritmo sinusal con ondas T aplanadas en caras lateral e inferior. La radiografía de tórax mostró una pequeña atelectasia en base pulmonar derecha y una lesión pleuropulmonar de aspecto inflamatorio en la base izquierda; esta lesión no aparecía en posteriores controles radiológicos. Se realizó un estudio ecocardiográfico, que fue normal, mostrando una fracción de eyección del 71%. La ecografía abdominal fue normal. En la TAC craneal sólo se apreciaban signos de atrofia cortical discreta. Se realizó un estudio electromiográfico y electroneurográfico, cuyos hallazgos fueron compatibles con un proceso miopático proximal afectando a miembros superiores e inferiores. Finalmente, se realizó una biopsia del músculo bíceps braquial derecho, cuyo informe fue el siguiente: al microscopio óptico (Fig. 1) se apreciaba una profusa infiltración grasa con acúmulos lipídicos de gran tamaño y con amplia variabilidad en el diámetro de las fibras, algunas de las cuales presentaba basofilia, observándose también algunas fibras hendidas. Existía aumento del colágeno intersticial; con técnicas oxidativas, se apreció aumento de la reacción, de predominio en regiones paranucleares y subsarcolémicas. En determinadas zonas aparecían muchas fibras con núcleos centrales. El estudio de inmunofluorescencia no mostró depósito alguno de inmunoglobulinas, complemento ni fibrinógeno. En el estudio ultraestructural (Fig. 2), aparecían abundantes glóbulos lipídicos en clara relación con mitocondrias, cuyo tamaño y número aparecía aumentado. El diagnóstico final fue de miopatía lipídica por déficit de carnitina, considerándose precisa la confirmación histoquímica. Por este motivo se realizó una medición simultánea de carnitina total en suero y músculo; la carnitina sérica mostró cifras normales de acuerdo a los standards de nuestro laboratorio de bioquímica clínica, mientras que la determinación de carnitina muscular mostró un valor de 0,4 μg/mg de tejido, considerado por debajo de la normalidad en nuestro laboratorio.
Se decidió comenzar a tratar a la paciente con carnitina oral, con lo que se consiguió cierta mejoría de carácter subjetivo, sin que la exploración ni los controles electromiográficos sufriesen modificaciones apreciables. Tras el alta hospitalaria, la paciente se mantuvo durante un año sin incidencias en su domicilio, al cabo del cual sufrió una nueva infección respiratoria que precisó ingreso hospitalario, desarrollándose una insuficiencia respiratoria progresiva que la condujo al exitus letalis.
DISCUSIÓN
La carnitina (1) (γ-trimetilamino-β-hidroxibutirato) está presente en muchos alimentos, pero los requerimientos orgánicos diarios sólo pueden conseguirse a través de una biosíntesis adicional. Sus dos precursores principales son la lisina y la metionina. Su síntesis implica la formación de ε-N-trimetil-lisina (2) y su desdoblamiento por oxidación en glicina y γ-trimetilaminobutirato; la β-hidroxilación de este último compuesto da lugar a la carnitina. En el hombre, todos los enzimas necesarios para la biosíntesis de carnitina se encuentran en el hígado, el riñón y el cerebro. El músculo esquelético y el cardíaco pueden participar también en su biosíntesis, pero carecen del enzima final, la γ-trimetilaminobutirato hidroxilasa. La carnitina sintetizada en hígado y riñón llega al resto de los tejidos a través de la circulación. Los niveles de carnitina en el músculo cardíaco y el esquelético son claramente superiores a los niveles séricos, y la captación de carnitina por parte de dichos tejidos se lleva a cabo mediante un mecanismo de transporte activo. Hasta el momento, se distinguen dos tipos principales de síndromes por deficiencia de carnitina: la forma predominantemente miopática y la forma sistémica (3).
La forma miopática de deficiencia de carnitina suele identificarse por la triada clásica de debilidad muscular progresiva con acúmulos lipídicos y déficit de carnitina en el músculo. Esta forma clínica es la que padecía nuestra paciente. La mayoría de estos casos cursa con niveles séricos normales de carnitina. En el tratamiento se ha utilizado prednisona, en algún caso asociándola a propranolol (4), pero los mejores resultados se han obtenido utilizando suplementos de carnitina (5) y dietas con bajo contenido graso (6) e incremento del contenido en carbohidratos, con resultados variables. Independientemente de la respuesta clínica al tratamiento, en ningún caso se ha conseguido incrementar los niveles musculares de carnitina. Hasta el momento, el caso de mayor edad publicado contaba 67 años en el momento del diagnóstico (7).
En cuanto a la forma sistémica del déficit de carnitina (8), se caracteriza clínicamente por episodios agudos de encefalopatía asociados con disfunción hepática, debilidad muscular progresiva predominantemente proximal y acúmulos lipídicos en el músculo, predominando en las fibras de tipo I, como ocurre en la forma miopática, y en otros tejidos del organismo. Los niveles de carnitina muscular suelen estar reducidos, como también suelen estarlo los niveles séricos y los de tejido hepático. Los ataques agudos suelen ir precedidos por vómitos, los cuales se siguen de un estado de confusión y estupor creciente que puede terminar en el coma. Entre los hallazgos de laboratorio más comunes, se encuentran la hipoglucemia, hipoprotrombinemia, hiperamoniemia y aumento de las enzimas hepáticas (9). Existe un deterioro del metabolismo de los ácidos grasos de cadena larga, ya que la carnitina es precisa para su oxidación (2,10). Durante los ataques agudos, la acidosis metabólica se da en el 50% de los casos. Los ataques pueden ser provocados por deprivación calórica. Los episodios encefalopáticos pueden ser similares a los del síndrome de Reye (11), pero en este trastorno, los niveles de carnitina sérica y tisular suelen estar normales, o sólo mínimamente reducidos. La enfermedad comienza a ser evidente en el primer año de vida, pero en ocasiones puede no ser diagnosticada hasta la adolescencia.
Recientemente se ha determinado que los estados de deficiencia primaria de carnitina constituyen un trastorno de herencia autosómica recesiva que afecta a la ruta oxidativa de los ácidos grasos, debido a un defecto en el transporte de carnitina. Se han identificado mutaciones que anulan la función de un transportador de carnitina y cationes denominado OCTN2 (12).
Debemos citar además otras situaciones que cursan con déficit de carnitina probablemente asociada a otras alteraciones metabólicas. Es el caso de algunos pacientes que siguen programa de hemodiálisis crónica (2), en los que se han descrito descenso de los niveles musculares de carnitina, como también se ha descrito en el suero y tejidos de pacientes con cirrosis hepática en estadío avanzado, y en los músculos de algunos pacientes con distrofia de Duchenne avanzada. La ausencia de síntomas tales como crisis eméticas o de disminución de nivel de conciencia en el caso que describimos, así como la inexistencia de alteraciones analíticas como hipoglucemias, acidemia o aciduria, además de los hallazgos anatomopatológicos, nos permiten descartar las acidurias orgánicas como causa de un posible déficit secundario de carnitina (13).
Un trastorno a diferenciar de los anteriores es el déficit de carnitina-palmitoil-transferasa (14,15), defecto de probable herencia autosómica recesiva con marcada reducción de su penetrancia para el sexo femenino. Sus síntomas comienzan de forma intermitente durante la primera o segunda década de la vida. Suelen aparecer dolor y debilidad muscular durante el ejercicio sostenido. Son clásicos los ataques recurrentes de mioglobinuria, que suelen ser consecutivos al ejercicio sostenido, especialmente si éste se realiza durante deprivación calórica o en exposición al frío. La insuficiencia renal puede complicar un ataque de mioglobinuria. Los enzimas musculares suelen incrementarse marcadamente durante las crisis, pero también pueden aumentar tras un ejercicio que no termine con mioglobinuria. Entre los ataques, los pacientes suelen tener fuerza muscular normal y no muestran anormalidades electromiográficas. La biopsia muscular no suele mostrar acúmulos lipídicos o sólo un ligero incremento de material graso, lo que constituye una clara diferencia con respecto a los estados de déficit de carnitina (4). Puede demostrarse este defecto enzimático en las biopsias musculares, así como en leucocitos y fibroblastos. En su tratamiento se han utilizado también dietas con bajo contenido graso que incluyen triglicéridos de cadena media, y alto contenido en carbohidratos, utilizando suplementos dietéticos antes y durante el ejercicio prolongado. Son trastornos clínicamente similares, pero de más reciente descripción, el déficit de piruvato-decarboxilasa (3) y el de acetil-coenzima A-deshidrogenasa (16). También se ha descrito una miopatía que cursa con déficit de carnitina y una alteración del metabolismo de los ácidos grasos de cadena larga asociada al uso de zidovudina (17).
Por último, la existencia de alteraciones oculares asociadas a patología muscular obliga a descartar las miopatías mitocondriales, ya que el cuadro clínico no reviste características miasteniformes. El caso que describimos no mostraba crisis de epilepsia mioclónica, episodios de focalidad neurológica central ni acidosis láctica, entre algunas de las características más significativas de estas miopatías; además, el trastorno ocular descrito en la exploración clínica era congénito y no progresivo y, como dato esencial, no se detectaron las características "fibras rasgadas" en la biopsia muscular (18).
Las miopatías lipídicas, a pesar de ser trastornos poco frecuentes, son entidades a tener en cuenta en el estudio de cualquier paciente con sintomatología miopática o elevación recurrente de las enzimas musculares, especialmente si consideramos que algunos de estos trastornos pueden pasar desapercibidos hasta la edad adulta y que en muchas ocasiones existen posibilidades terapéuticas.
Bibliografía
1. Bieber LL. Carnitine. Ann Rev Biochem 1988; 57: 261-83. [ Links ]
2. Gilbert EF. Carnitine deficiency. Pathology 1985; 17 (2): 161-71. [ Links ]
3. Angelini C. Lipid storage myopathies,. A review of metabolic defect and of treatment. J Neurol 1976; 214 (1): 1-11. [ Links ]
4. Isaacs H, Heffron JJA, Badenhorst M, Pickering A. Weakness associated with the pathological presence of lipid in skeletal muscle: a detailed study of a patient with carnitine deficiency. J Neurol Neurosurg Psychiatry 1976; 39 (11): 1114-23. [ Links ]
5. Levitan MD, Murphy JT, Sherwood WG, Deck J, Sawa JM. Adult onset systemic carnitine deficiency: favorable response to L-carnitine supplementation. Can J Neurol Sci 1987; 14 (1): 50-4. [ Links ]
6. Snyder TM, Little BW, Roman-Campos G, McQuillen JB. Succesful treatment of familial idiopathic lipid storage myopathy with L-carnitine and modified lipid diet. Neurology 1982; 32 (10): 1106-15. [ Links ]
7. Martin JJ, Vercruyssen A, de Barsy T, Ceuterick C. Muscle carnitine deficiency in old age. Case report and therapeutic results. Clin Neurol Neurosurg 1985; 87 (4): 275-81. [ Links ]
8. Karpati G, Carpenter S, Engel AG, Watters G, Allen J, Rothman S, et al. The syndrome of systemic carnitine deficiency. Clinical, morphologic, biochemical and pathophysiologic features. Neurology 1975; 25 (1): 16-24. [ Links ]
9. Breningstall GN. Carnitine deficiency syndromes. Pediatr Neurol 1990; 6 (2): 75-81. [ Links ]
10. Scholte HR, Luyt-Houwen IE, Vaandrager-Verduin MH. The role of the carnitine system in myocardial fatty acid oxidation: carnitine deficiency, failing mitochondria and cardiomyopathy. Basic Res Cardiol 1987; 82 Suppl 1: 63-73. [ Links ]
11. Chapoy PR, Angelini C, Brown WJ, Stiff JE, Shug AL, Cederbaum SD. Systemic carnitine deficiency:a treatable inherited lipid-storage disease presenting as Reye´s syndrome. N Engl J Med 1980; 303 (24): 1389-94. [ Links ]
12. Wang Y, Kelly MA, Cowan TM, Longo N. A missense mutation in the OCTN2 gene associated with residual carnitine transport activity. Hum Mutat 2000; 15 (3): 238-45. [ Links ]
13. Ribes Rubio A, Rodés Monegal M. Alteraciones del metabolismo de los aminoácidos y de los ácidos orgánicos. En: Farreras P, Rozman C, editores. Medicina Interna. 13ª ed. Madrid: Harcourt Brace; 1997, p. 1867-71. [ Links ]
14. Bank WJ, DiMauro S, Bonilla E, Capuzzi DM, Rowland LP. A disorder of muscle lipid metabolism and myoglobinuria: absence of carnitine palmityl transferase. N Eng J Med 1975; 292: 443-9. [ Links ]
15. Reza MJ, Kar NC, Pearson CM, Kark RAP. Recurrent Myoglobinuria due to muscle carnitine palmityl transferase deficiency. Ann Int Med 1978; 88: 610-5. [ Links ]
16. Carroll JE. Myopathies caused by disorders of lipid metabolism. Neurol Clin 1988; 6 (3): 563-74. [ Links ]
17. Dalakas MC, Leon-Monzon ME, Bernardini I, Gahl Wa, Jay CA. Zidovudine-induced mitochondrial myopathy is associated with muscle carnitine deficiency and lipid storage. Ann Neurol 1994; 35--84-9: 482-7. [ Links ]
18. Urbano-Márquez A, Grau Junyent JM, Casademont Pou J, Cardellach López F. Enfermedades musculares. En: Farreras P, Rozman C, editores. Medicina Interna. 13ª ed. Madrid: Harcourt Brace; 1997. p. 1573. [ Links ]

