










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkAnales de Medicina Interna
Print version ISSN 0212-7199
An. Med. Interna (Madrid) vol.20 n.6 Jun. 2003
| Actualización de la cistinuria: aspectos clínicos, bioquímicos y genéticos J. A. ORTS COSTA, A. ZÚÑIGA CABRERA1, J. MARTÍNEZ DE LA CÁMARA Y SALMERÓN2 Área de Diagnóstico Biológico (Bioquímica, 1Biología Molecular). Hospital de la Ribera. Alzira, Valencia. 2Urolitolab. Benidorm, Alicante
|
| CYSTINURIA UPDATING: CLINICAL, BIOCHEMICAL AND GENETIC ASPECTS
| ||
| RESUMEN La cistinuria es una enfermedad autosómica recesiva, con una incidencia estimada de un caso por cada 7000 nacidos vivos, que ocasiona una elevada excreción urinaria de cistina, lisina, arginina, y ornitina. Descrito ya en 1908, por sir Archibald Edward Garrod, como uno de los cuatro primeros errores del metabolismo conocidos, se caracteriza por un defecto en el transporte de cistina y de los aminoácidos dibásicos que afecta a su reabsorción en el túbulo renal y tracto gastrointestinal. Hasta la fecha, según hallazgos moleculares recientes, se han identificado dos genes como responsables de esta enfermedad: SLC3A1 y SLC7A9. Una correcta identificación fenotípica y/o genotípica de los pacientes cistinúricos permitirá una mejor profilaxis y terapia para esta patología. La cistinuria ocasiona urolitiasis recurrente (aproximadamente el 1-2 % de los cálculos renales del adulto) debida a una deficiente solubilidad de la cistina a pH bajo. La calcinogénesis y sus complicaciones asociadas son las únicas manifestaciones clínicas de esta enfermedad. Las medidas profilácticas, basadas en una alta ingesta hídrica y la alcalinización de la orina con citrato potásico, en primera línea; y la utilización del arsenal farmacológico de derivados tiólicos, en segunda línea, se deberá extremar en aquellos pacientes (como los homocigotos tipo I) de alto riesgo litógeno. No obstante, para aproximadamente un 50 % de los pacientes sometidos a control preventivo y que, a pesar de ello, desarrollarán litiasis recidivante, los procedimientos urológicos y quirúrgicos serán la única alternativa posible. En este artículo revisamos y actualizamos el conocimiento sobre los aspectos bioquímicos, genéticos, clínicos, diagnósticos, de prevención, tratamiento y pronósticos de esta, relativamente rara, enfermedad. | ABSTRACT Cystinuria is an autosomal recessive disorder with an estimated incidence of 1 case in 7000 live births, that results in elevated urinary excretion of cystine and dibasic aminoacids: ornithine, lysine and arginine. Discussed by Sir Archibald Edward Garrod, in 1908, as one of the four first known inborn errors of metabolism, it is characterized by a defect in transport of cystine and dibasic aminoacids, that affects their reabsortion in both renal tubule and gastrointestinal tract. To date, according to the recent molecular findings, two genes have been identified as responsible for this disease: SLC3A1 and SLC7A9. A more accurate pheno/genotyping identification of cystinuric patients will allow to improve prophilaxis and therapy for this illness. Cystinuria only causes recurrent urolithiasis (about 1-2 % of renal calculi in adults) and its associated complications as clinical feature because of poor cystine solubility at low pH. An accurate control over prohylaxis (based on high water intake and potassium citrate treatment, on first line, and tiol-derivatives treatment, on second line) must be taken in patients -like homozygous type I- with high lithiasis risk. However, approximately one half of patients under prophylaxis control will develop recurrent lithiasis; in this case, only urology or surgical approaches would be possible. Updated knowledge about biochemical, genetic, clinical, diagnosis, prevention, treatment and prognosis aspects of this, relatively unusual, disease has been reviewed in this article. |
Orts Costa JA, Zúñiga Cabrera A, Martínez de la Cámara y Salmerón J. Actualización de la cistinuria: aspectos clínicos, bioquímicos y genéticos. An Med Interna (Madrid) 2003; 20: 317-326.
Trabajo aceptado: 27 de marzo de 2003
Correspondencia: J. Antonio Orts Costa. Área de Diagnóstico Biológico. Hospital de la Ribera. Ctra. Alzira-Corbera, km. 1. 46600 Alzira (Valencia). e-mail: jaorts@hospital-ribera.com
INTRODUCCIÓN
Es sorprendente que una enfermedad rara, con una incidencia aproximada en neonatos de 1/7.000, fuera descrita tan remotamente como a principios del siglo XIX e incluída, con insólita presteza –al principio del siglo XX– entre los primeros errores congénitos del metabolismo conocidos. En 1810, Wollaston, observó unos cálculos diferentes a los usuales, compuestos por placas hexagonales y solubles en álcali, pensó, erróneamente, que eran exclusivamente de origen vesical –recordemos que en la era preantibiótica la mayoría de los cálculos eran consecuencia de infecciones urinarias recidivantes de vías bajas, probablemente de estruvita y urato de amonio, en su mayoría– y los supuso constituidos por un compuesto que él denominó “óxido cístico” (kystis significaba vejiga en griego) (1). Marcet, en 1817, mostró que este tipo de litiasis podía presentarse, también, en el riñón (2). Poco tiempo después, en 1824, Stromeyer identificó en la orina de pacientes afectos los típicos cristales que han servido –y siguen sirviendo– para el diagnóstico de la enfermedad (3). En 1830, Berzelius, se percató de que los cristales estaban compuestos por aminas y los renombró como “cistina” (4). Finalmente, en 1851, Toel confirmó la hipótesis de Prout en 1823 de que la enfermedad era ocasionada por una excreción urinaria excesiva de cistina (5,6).
Garrod, en 1908, los incluyó dentro de los cuatro primeros errores congénitos del metabolismo conocidos suponiendo, también equivocadamente, que se trataba de un defecto enzimático, pese a que a finales del anterior siglo, Von Udransky y Baumann, presumieron una alteración intestinal en el transporte de aminoácidos (7,8). Hasta 1947, no se comprobó que estaba asociada a una excreción urinaria excesiva de tres aminoácidos: arginina, lisina, y ornitina (9,10). La hipótesis de que era causada por un defecto en la reabsorción renal e intestinal de estos aminoácidos y la cistina, formulada por Dent y Rose en 1951, fue confirmada en 1961 por Milne (11,12). La primera descripción del patrón hereditario fue realizada por Harris en 1955 (13).
Se trata de una enfermedad hereditaria en la que, como consecuencia de mutaciones en los genes SLC3A1 (ubicado en el cromosoma 2, 2p16.3-21) y SLC7A9 (situado en el cromosoma 19, 19q12-13), se ve afectado el transporte, a nivel tubular renal y en algunos casos intestinal, de determinados aminoácidos dibásicos: arginina, lisina, ornitina, así como el de la cistina (dímero del aminoácido cisteína) (Fig. 1). Por tanto, cursa con una hiperexcreción urinaria de cistina y de los aminoácidos citados, siendo la cistina la que se sobresatura en la orina, precipitando, especialmente en medio ácido, y formando unos cálculos duros, radiopacos, de aspecto acaramelado y difícil fragmentación con litotricia extracorpórea con ondas de choque (LEOC) (14).
El diagnóstico definitivo se realiza según a la información aportada por el laboratorio mediante la determinación de la composición exacta de los cálculos, por constatación de la hipercistinuria o bien, y gracias a la biología molecular, por caracterización del gen implicado.
No existe más clínica asociada con esta enfermedad que la relacionada con los procesos litiásicos, siendo el tratamiento y la profilaxis más complejos que en el resto de litiasis.
No hay que confundir la cistinuria (OMIM 220100 y 600918) con la cistinosis (OMIM 219800), una rara enfermedad multiorgánica que se caracteriza por depósitos lisosomales de cristales de cistina. Esta última, de transmisión autosómica recesiva, y con una baja incidencia (1/ 100 000-200 000 nacidos vivos), está ocasionada por mutaciones del gen CTNS, ubicado en el cromosoma 17p13 e implicado en el transporte de cistina por la membrana lisosomal. Cursa, principalmente, con síndrome de Fanconi (fallo tubular en la reabsorción de pequeñas moléculas como los aminoácidos) causado por nefropatía por acúmulo de cristales de cistina en las células tubulares. Presenta, hipercistinuria e hiperaminoaciduria (además de hiperfosfaturia, proteinuria, hipercalciuria, etc), sin embargo y al igual que las otras enfermedades afectadas de síndrome de Fanconi (Wilson, síndrome de Lowe, galactosemia y enfermedades de almacenamiento del glucógeno), no llegan a formar cálculos de cistina como sucede en la cistinuria. La afectación sistémica, la presencia de un precoz síndrome de Fanconi y la ausencia de litiasis obvian la necesidad de un diagnóstico diferencial con la cistinuria, debiéndose descartar, más bien, un síndrome de Bartter, o una diabetes insípida (15) .
En el presente trabajo nos proponemos revisar los aspectos fisiopatológicos, genéticos, clínicos, diagnósticos, de prevención, tratamiento y pronóstico de la cistinuria.
FISIOPATOLOGÍA DEL TRANSPORTE RENAL E INTESTINAL DE AMINOÁCIDOS
En el riñón, los aminoácidos son ultrafiltrados, a través del glomérulo renal, al fluido tubular, posteriormente son reabsorbidos en más de un 98 % a nivel de túbulo contorneado proximal, gracias a mecanismos de transporte activo (contra un gradiente electroquímico) que depende del gradiente de sodio luz-tubular/célula tubular y de la energía intracelular almacenada. En adultos, el 99 % de la cistina filtrada es reabsorbida, aunque la contribución relativa del túbulo contorneado proximal con respecto al distal es incierta. En pacientes cistinúricos, la excreción fraccional de cistina es el 100 % de la filtrada y en algunos casos la supera, lo que implica algún tipo de secreción (14).
Investigaciones con animales de experimentación han demostrado que los aminoácidos dibásicos inhiben competitivamente la reabsorción de cistina a nivel tubular, provocando una hipercistinuria inducida. Estudios en riñón de rata han demostrado que la cistina es reducida intracelularmente al monómero cisteína mediante la glutation-cisteína transhidrogenasa, siendo la cisteína la principal forma intracelular (Fig. 1). Diferentes investigaciones in vivo han definido múltiples mecanismos renales de reabsorción para la cistina y los aminoácidos dibásicos. Así, en los pacientes cistinúricos se han encontrado, al menos, dos sistemas de reabsorción: uno de baja capacidad y alta afinidad (baja Km) que es el defectivo en la cistinuria, y otro con alta capacidad y baja afinidad que permanece inalterado en la mencionada patología.
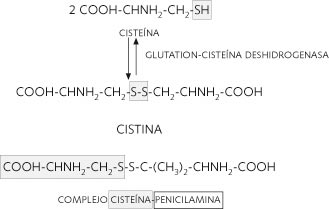
Fig. 1.
Mediante estudios de microperfusión in vitro en vesículas de membrana de las microvellosidades de los túbulos proximales del riñón y las del intestino delgado en rata, se ha comprobado que tanto la cistina como los aminoácidos dibásicos comparten el mismo sistema de transporte de alta afinidad localizado, en el caso del riñón, en el túbulo recto proximal o pars recta (S 3). Este sistema de transporte (b0,+), compartido, igualmente, con aminoácidos dibásicos, se encuentra en la superficie luminal de las células epiteliales renales e intestinales. Perteneciente a los transportadores heteroméricos, está constituido por dos polipéptidos: una subunidad pesada y una ligera unidas por puente disulfuro. La subunidad pesada está formada por glicoproteínas N-glicosiladas tipo II de membrana, mientras que la subunidad ligera, lo está por proteínas no glicosiladas de membrana. La proteína perteneciente al primer grupo rBAT está implicada en un sistema de reabsorción de alta afinidad para la cistina en el túbulo recto proximal de la nefrona y en el tracto intestinal (14).
Por tanto, podemos definir a la cistinuria como un defecto hereditario en el transporte de alta afinidad para la cistina, compartido con aminoácidos dibásicos, a través de células epiteliales del túbulo renal y enterocitos yeyunales.
Las mutaciones en los genes SLC3A1 y SLC7A9 (según nomenclatura de la Base de Datos del Genoma, Genome Data Base), que codifican el sistema transportador b0,+, son las responsables de la cistinuria tipo I y tipo no I, respectivamente. El mecanismo de captación activado por estos genes está siendo estudiado intensivamente en ovocitos de rana y celulas renales epiteliales cultivadas, y recuerda a los sistemas de transporte estudiados en blastocitos de rata (16).
Ha sido comprobado que los pacientes homocigotos fenotípicos excretan más de 113 mmol cistina/mmol creatinina, considerándose esta concentración como generadora de litiasis (17).
Inicialmente, los pacientes heterocigotos fueron clasificados fenotípicamente por Rosenberg (18) en 1966 como pertenecientes a tres tipos diferentes de acuerdo con la excrección diaria de cistina: I, II y III. Así:
—Tipo I (portadores silentes), indistinguibles de los sujetos normales
—Tipo II (excreción moderada).
—Tipo III (excreción ligera, en este tipo la captación intestinal de cistina no se encuentra completamente alterada).
Posteriormente (17,19), los cistinúricos han sido reclasificados fenotípicamente en:
—Tipo I, forma recesiva completa con los padres y familiares del afecto (heterocigotos recesivos “completos”) con aminoaciduria dentro del rango de referencia (<420 mmol/ 24 h).
—Tipo no I, forma recesiva “incompleta” o co-dominantes), cuyos padres y familiares presentan aminoacidura con alguno de los aminoacidos implicados con excreción por encima del límite superior del rango de referencia (>420 mmol/ 24 h).
El mejor discriminador para distinguir a los heterocigotos tipo no I de la población normal es la excreción urinaria de lisina/creatinina (17). Los homocigotos y heterocigotos tipo no I pueden ser diferenciados gracias a que los primeros excretan en orina grandes cantidades de cistina y aminoácidos dibásicos y los últimos una mayor relación: lisina+cistina/arginina+ornitina (20). La absorción de lisina y arginina tras sobrecarga oral de las mismas nos ayuda a distinguir entre los diversos fenotipos (21) (Tabla I). Recientemente se ha descrito una nueva clasificación genotípica tipo A (debido a dos mutaciones en el gen SLC3A1), tipo B (a dos mutaciones en el SLC7A9) y AB en ambos genes (19).
Durante el primer año de vida, el sistema de transporte renal de aminoácidos permanece inmaduro, incrementándose la hiperexcreción de cistinuria, pudiendo los heterocigotos simular una excreción análoga a la de los homocigotos (14).
A pesar de la evidencia de un defecto intestinal en el transporte de aminoácidos, no ha sido descrita clínica alguna de diarrea o síntomas de malabsorción que afecten a los pacientes cistinúricos. Una explicación posible a este fenómeno es que la presencia de transportadores de baja afinidad permitan una suficiente absorción de cistina y aminoácidos dibásicos en el tracto intestinal; otra justificación sería la posible absorción de dipéptidos, gracias a transportadores de oligopéptidos (que se han localizado en las membranas apicales de los enterocitos), y que estos dipéptidos, una vez en el enterocito sufriesen una hidrólisis a simples aminoácidos que serían vertidos al torrente circulatorio (14).

GENÉTICA
El estudio de las bases moleculares de la cistinuria ha permitido caracterizar una familia de transportadores heteroméricos de aminoácidos (HTAs; heteromeric amino-acid transporters) de la membrana plasmática de las células de mamíferos. Hasta la fecha, los HTAs son la única familia de estos transportadores con una naturaleza heteromérica, y están formados por una cadena pesada (HSHAT) y una cadena ligera (LSHAT). Las alteraciones en estos transportadores son las responsables no sólo de la cistinuria sino también de otra enfermedad conocida como intolerancia a proteínas con lisinuria (LSI: OMIM 222700) (22,23). En el caso de la cistinuria, el sistema de transporte de aminoácidos implicado se conoce como b0,+, y está formado por una subunidad pesada, denominada rBAT, y una subunidad ligera, identificada como b0,+AT, codificadas por los genes SLC3A1 y SLC7A9 respectivamente (24). El papel de rBAT en la cistinuria tipo I se demostró en 1994 (25), mientras que el de b0,+ AT en la cistinuria tipo no I se confirmó en 1999 (14) (Tabla II).
Tradicionalmente, se han distinguido tres tipos de cistinuria: tipo I, II y III. Sin embargo, esta clasificación no se correlaciona bien con los hallazgos a nivel molecular, por lo que, actualmente, se clasifican como tipo I (OMIM 220100) y tipo no I (OMIM 600918; y que clínicamente se subdivide en tipo II y tipo III). La cistinuria de tipo I y tipo no I se distingue en base a la aminoacidouria de cisteína y de aminoácidos dibásicos de los heterocigotos obligados (26) (Tabla II). Los heterocigotos de tipo I son silentes, mientras que en los heterocigotos de tipo no I hay un grado variable de hiperexcreción urinaria de estos aminoácidos, siendo mucho mayor en los de tipo II que en los de tipo III.
También, han sido descritos pacientes con un tipo mixto que heredan alelos de tipo I y de tipo no I de sus progenitores (27). La cistinuria de tipo I es la mayoritaria, y representa, aproximadamente, el 60% de todos los casos de esta enfermedad.

CISTINURIA TIPO I
Mediante análisis de ligamiento del ADN de familias con cistinuria, se localizó el gen defectivo en el cromosoma 2 (28). Este gen, se denominó en un principio como rBAT (related to b0,+ amino acid transporter), y actualmente se conoce como SLC3A1 (SLC de solute carrier) en la nomenclatura de la Base de Datos del Genoma. Algunos autores, siguen utilizando la designación de rBAT para genes similares que codifican transportadores para aminoácios básicos en otros sistemas, así como para las proteínas que estos genes codifican.
El gen SLC3A1, codifica la subunidad pesada (rBAT) del sistema b0,+ de transporte renal de la cistina y de los aminoácidos dibásicos lisina, arginina y ornitina. Este gen está implicado en la patogénesis de la cistinuria tipo I con carácter autosómico recesivo. Se encuentra localizado en el brazo corto del cromosoma 2 (2p16.3-p21) (29,30) . Su organización genómica fue establecida en 1996 (31,32). El gen tiene una longitud aproximada de 45 kb y contiene 10 exones. El tamaño de los intrones oscila entre los 500 pb (intrón 2) y los 13.000 pb (intrón 4). Todas las uniones intrón/exón presentan las secuencias 5’ GT para el aceptor y 3’ AG para el dador, típicas de los eucariotas. La región del promotor ha sido analizada y se ha localizado la caja TATA a 98 pb corriente arriba (upstream) del primer codón ATG. La proteína sintetizada (rBAT) es una glicoproteína de membrana tipo II con un único dominio transmembranal con el extremo amino-terminal intracelular y el extremo carboxi-terminal extracelular. La poteína rBAT se expresa únicamente en el túbulo proximal del riñón y en las células intestinales con borde en cepillo.
Una vez que se estableció la estructura genómica de SLC3A1, el estudio en distintas poblaciones de las familias con cistinuria ha permitido describir más de 60 mutaciones causantes de la enfermedad. La naturaleza de las mutaciones es muy variable e incluye: mutaciones sin sentido, codones de parada, errores en la maduración del ARN, delecciones e inserciones (Tabla III). Todas causan, por diversos mecanismos, una expresión incorrecta de la proteína transportadora en la membrana de las células renales (33). La gran mayoría de las mutaciones son específicas de una población, lo que sugiere un origen reciente o su circunscripción a poblaciones genéticamente aisladas. Una de las más frecuentes y que ha sido descrita principalmente en las poblaciones mediterráneas es la M467T, causada por el cambio en el nucleótido 1.400 de T → C, que origina que el codón 467 codifique para una metionina en vez de una treonina (34). Estudios realizados sobre la patogeneicidad de la mutación M467T indican que esta causaría un enlentecimiento del tráfico de la proteína desde el retículo endoplasmático hasta la membrana celular (35). Otras con una distribución amplia son: R270X, E483X, T216M y 1500 +1G → T.
De los estudios diseñados para correlacionar genotipos anormales específicos con la presentación fenotípica, es concluyente que los cistinúricos de tipo I presentan mutaciones en ambos alelos para el gen SLC3A1. Las dos mutaciones que un paciente hereda pueden o no ser las mismas; una mutación distinta del mismo gen podría ser heredada de cada uno de los progenitores dando origen a la enfermedad (36) .
CISTINURIA TIPO NO I
Desde el descubrimiento del gen SLC3A1, las evidencias clínicas y fisiológicas ya apuntaban a un origen genético heterogéneo de la cistinuria, que explicara el tipo no I de carácter codominante o incompletamente recesivo. Los primeros datos de un locus distinto fueron aportados por Bisceglia y cols. (37) que lo situaban en el brazo largo del cromosoma 19 (19q13.1). En 1999, el Consorcio Internacional de Cistinuria (38) identificó al gen SLC7A9 como responsable de la cistinuria tipo no I. Este gen codifica una proteína denominada b0,+AT, que pertenece a la familia de las subunidades ligeras de los transportadores de aminoácidos de la familia HATS. b0,+AT es una proteína no glicosilada, con 12 dominios transmembranales y con los extremo amino y carboxi-terminales intracelulares. Al parecer, interacciona con la subunidad pesada rBAT del sistema b0,+ mediante un puente disulfuro, pero se desconoce hasta la fecha si forman un heterodímero o tienen un nivel más complejo de oligomerización.
Hasta la fecha, se han identificado más de 35 mutaciones diferentes del gen SLC7A9 en distintas poblaciones siendo, como en el caso del gen SLC3A1, muy variadas (Tabla IV). Al parecer, las mutaciones que afectan a aminoácidos con cadenas laterales pequeñas localizados en los dominios transmembranales se asocian con un fenotipo más severo. Estos estudios no han permitido diferenciar claramente los fenotipos tipo II y tipo III, y al parecer, estos tipos junto con algunos casos de tipo I, serían la manifestación de diferentes variantes alélicas del gen SLC7A9, estando probablemente implicados otros factores genéticos y medioambientales.

El Consorcio Internacional de Cistinuria (39) ha propuesto dos hipótesis que explicarían el carácter recesivo de las mutaciones del gen SLC3A1 y el carácter codominante o incompletamente recesivo del gen SLC7A9. La primera hipótesis, presupone que el sistema transportador b0,+ está constituido por más de una subunidad rBAT y b0,+AT; un alelo mutado de la cadena pesada rBAT provocaría un defecto en el tráfico de la proteína mientras que un alelo mutante de la cadena ligera produciría un efecto dominante. La segunda hipótesis, considera que la cadena ligera se asocia con otra proteína, además de rBAT, y daría lugar a un transporte de cistina en un segmento tubular proximal diferente. Este segundo mecanismo aún no se conoce a nivel molecular, pero si el gen SLC7A9 también participara en él, explicaría el fenotipo incompletamente recesivo de la cistinuria no tipo I.
DIAGNÓSTICO
Es de manifiesta utilidad para el cribado de pacientes cistinúricos la utilización de la reacción de Brand (cianuro-nitroprusiato), una prueba semicuantitativa a que sometemos la orina de los pacientes sospechosos. Cuando el cianuro sódico se mezcla con orina alcalinizada , la cistina es reducida a cisteína que en presencia de nitroprusiato sódico da un color magenta característico. Es positiva en pacientes con cistinuria superior a 315 mM, por lo tanto detecta los pacientes litógénicos aunque no todos los heterocigotos. El test de Brand no ofrece la máxima sensibilidad ni especificidad diagnósticas, dando tanto falsos negativos, como falsos positivos en pacientes homocistinúricos y acetonúricos (40,41). Kutler propuso una modificación de esta reacción mediante reactivos en polvo a los que se añadía la orina y que, quizá, es más sencilla de realizar.
Otras técnicas químicas para la detección de cistina son: la de Sullivan, semicuantitativa, específica de la cistina frente a la homocisteína (en esta reacción química la cistina una vez transformada en cisteína reacciona con la 1, 2-naftoquinona-4-sulfonato sódico dando color marrón); y la de Shinohara y Padis, técnica cuantitativa (en la que la cistina reducida a cisteína mediante bisulfito sódico reacciona con ácido fosfotúngstico) (42) .
El procedimiento diagnóstico más simple, aunque sólo demostrable en el 19-26 % de los pacientes homocigotos, es la identificación microscópica en el sedimento urinario de los típicos cristales hexagonales planos, ya sean aislados o bien formando maclas. Son más fácilmente observables en la primera orina de la mañana, ya que es más ácida y concentrada; en cualquier caso si acidificamos la orina con ácido acético conseguiremos la precipitación de cristales que no son visibles con orina fresca.
El aspecto macroscópico que presentan los cálculos puros de cistina, observado mediante microscopía estereoscópica (lupa binocular), es de color miel-caramelo, céreo, brillante muy duro; de morfología y tamaño variable (aunque generalmente redondeada y de gran tamaño) (Fig. 2). Su estructura microcristalina se puede caracterizar mediante: microscopía diferencial de polarización (Fig. 3), microscopía electrónica de barrido (Fig. 4) y microanálisis por energía dispersiva de rayos X, siendo diagnósticas las formas prismáticas hexagonales, generalmente en maclas.
Los cristales de ácido úrico anhidro también presentan estructura hexagonal (43).

Fig. 2. Cálculo de cistina observado mediante microscopía
esteroscópica (lupa binocular). De aspecto acaramelado y brillante.
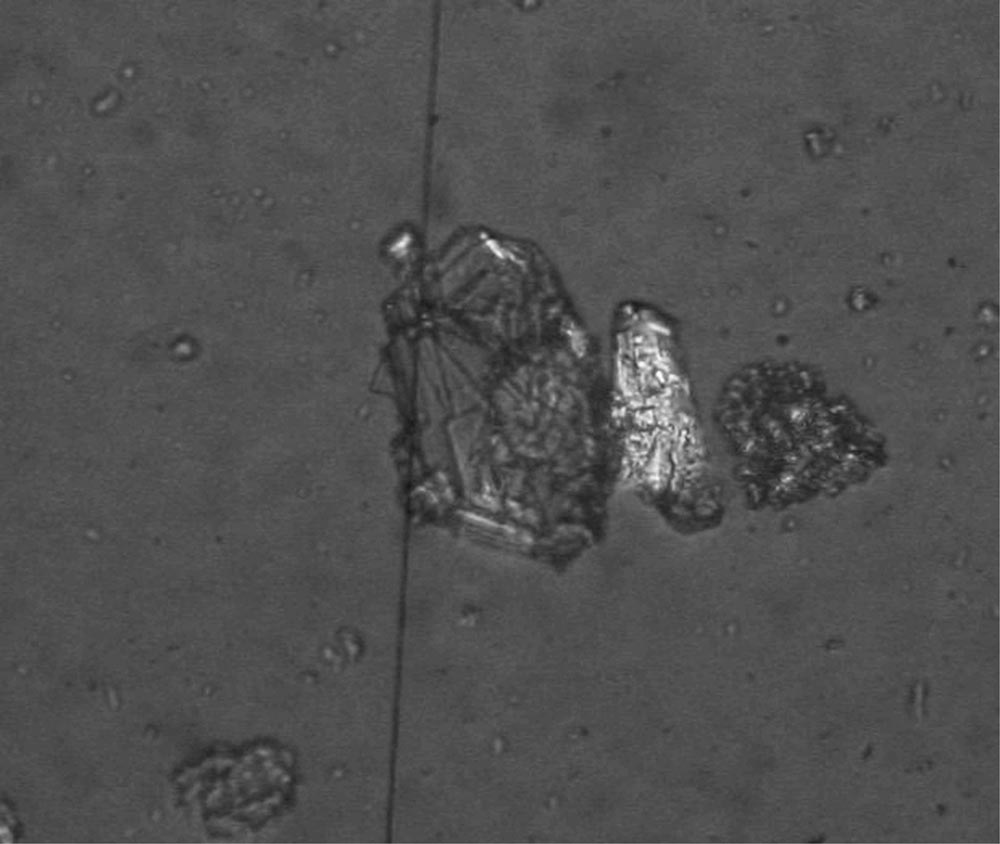
Fig. 3. Cristales de cistina, procedentes de un cálculo, observados
mediante microscopía de polarización diferencial. Se observa
perfectamente la estructura microcristalina hexagoinal.

Fig. 4. Cristales de cistina, observados mediante microscopía
electrónica de barrido. Destaca tridimensionalmente la estructura
microcristalina hexagonal en maclas, propia de los cálculos de
cistina (Fotografía obtenida en el Laboratorio d'Investigación
en Litiasi Renal, Prof. Dr. Félix GRases Freixedas.
Mallorca. Universitat de les Illes Balears).
Es muy importante la distinción microscópica entre los cristales hexagonales de cistina y los de ácido úrico anhidro. En el sedimento urinario los cristales son de ácido úrico anhidro suelen ser amarillentos, sin embargo los de cistina son incoloros. Si se observan bajo microscopía diferencial de polarización los cristales aislados y no maclados de cistina no polarizan la luz pese a ser anisótropos (comportamiento isotrópico) y esta propiedad nos permite diferenciarlos de los de ácido úrico anhidro que sí polarizan (anisótropos). La simetría regular y los ángulos próximos a un hexágono perfecto, en el caso de la cistina, nos ayudarán, igualmente, a distinguirlos. Sí la muestra procede de un cálculo se observarán, mediante microscopía de polarización, cristales maclados que polarizan pero con colores de interferencia bajos (grises) al contrario que los de ácido úrico que son mucho más altos y vivos, los ángulos y simetrías permiten, igualmente, diferenciarlos.
Un 50 % de los cálculos se asocian a oxalato cálcico, estruvita y ácido úrico.
Mediante espectrofotometría infrarroja se obtiene un espectro característico y diganóstico de la composición del cálculo, presentando múltiples bandas de tensión entre 1600 y 625 cm-1 (44) .
Se ha utilizado la cromatografía en capa fina como cribado de la enfermedad, aunque no es muy eficaz porque las sales urinarias tienden a crear malos desarrollos, especialmente de la cistina (42,45). La cromatografía de intercambio iónico es útil para cuantificar la cistinuria (46). Aunque actualmente la metodología que mejor caracteriza fenotípicamente esta enfermedad es la cuantificación de la cistina y/o aminoácidos dibásicos en orina por cromatografía líquida de alta resolución (HPLC) con derivatización de precolumna, considerándose un paciente fenotípicamente cistinúrico cuando la excreción urinaria de al menos uno de los aminoácidos cistina, lisina, arginina u ornitina, así como la suma total de la excreción total de los mencionados aminoácidos supera el límite superior de los valores de referencia (17,47). La cromatografía líquida-espectrometría de masas también se ha utilizado con la misma finalidad (48).
El análisis genético, aunque definitivo en casos individuales, no parece ser de utilidad en el cribado de la cistinuria, dado la baja frecuencia de mutaciones (baja prevalencia de la enfermedad) y la variabilidad de éstas (49,50).
CLÍNICA, PREVENCIÓN Y TRATAMIENTO
Las litiasis cistínicas constituyen sólo el 1-3 % de todas las litiasis en los adultos y sobre un 6-8% en los niños. La incidencia de esta enfermedad varía entre 1/15.000 encontrada en EEUU ,1/2500 en los judíos israelitas de origen libanés o 1/1887 en la Comunidad Valenciana (14,17,51). Las cifras de incidencia dependerán del criterio establecido para diagnosticar la cistinuria y del método de cribado utilizado, probablemente la mayoría de las estimaciones basadas en cribados neonatales están sobrevaloradas, dado la mayor excreción de cistina por inmadurez del túbulo renal en recién nacidos. Lógicamente, al tratarse de una enfermedad de transmisión hereditaria es más frecuente en comunidades con alto grado de cosanguinidad. La tasa de litiasis en pacientes cistinúricos no se conoce ya que no todos los pacientes cistinúricos desarrollan litiasis, ésta parece estar influenciada por diversos factores: la excreción de inhibidores de la calculogénesis, ambientales, o el balance salino.
Los característicos cristales hexagonales y planos pueden ser observados en prácticamente todos los sedimentos urinarios (preferiblemente de primera hora de la mañana porque la orina tiende a ser más concentrada y ácida) de los pacientes afectos. El debut de la sintomatología litiásica puede observarse, indistintamente, en la primera década de la vida (25-30%), adolescencia (25-30 %) o en los adultos, aunque el pico de mayor frecuencia se sitúa en la tercera década de la vida. Afecta por igual a ambos sexos (52). La edad de presentación de la litiasis parece depender del tipo de cistinuria, en un estudio realizado en Quebec más del 50% de pacientes de tipo I no tratados padecieron el primer episodio en la primera década de vida, sin embargo no ocurrió esto en los de tipo no I (36).
Como el defecto en el transporte intestinal carece de consecuencias clínicas, debido a su compensación por la absorción de los aminoácidos en forma dipeptídica (53), la clínica de la cistinuria es exclusivamente la nefro-urológica por litiasis.
Se describió por Scriver, en 1970, una mayor prevalencia de enfermedades mentales en pacientes cistinúricos, sin embargo esta asociación no ha sido corroborada posteriormente (54).
Clínicamente, se asocia una mayor excreción urinaria de cistina y litogénesis con los fenotipos homocigotos, aunque no todos los homocigotos son litógenos, siendo el homocigoto tipo I el que presenta la litiasis más precozmente. Los heterocigotos tipo no I tienen menor excreción de cistina y la litogénesis y su frecuencia son menores (36,47).
Los cálculos de cistina aunque son de tamaño y morfología variables (generalmente redondeados), a veces, adquieren tamaños desproporcionados, son radiopacos (Fig. 5), y en ocasiones se recubren, al infectarse, de una capa de fosfocarbonato o fosfato amónico-magnésico ( en este caso la evolución es similar a la de los cálculos coraliformes). La incidencia de litiasis es similar en ambos sexos (42) y es particularmente recidivante dado su carácter genético (14).
La presentación de la enfermedad es la clásica de la litiasis: hematuria micro o macroscópica, cólico nefrítico (con o sin expulsión del cálculo) y dolor lumbar. Puede acompañarse de infección urinaria, piuria estéril, obstrucción de vías urinarias y, eventualmente, fallo renal.
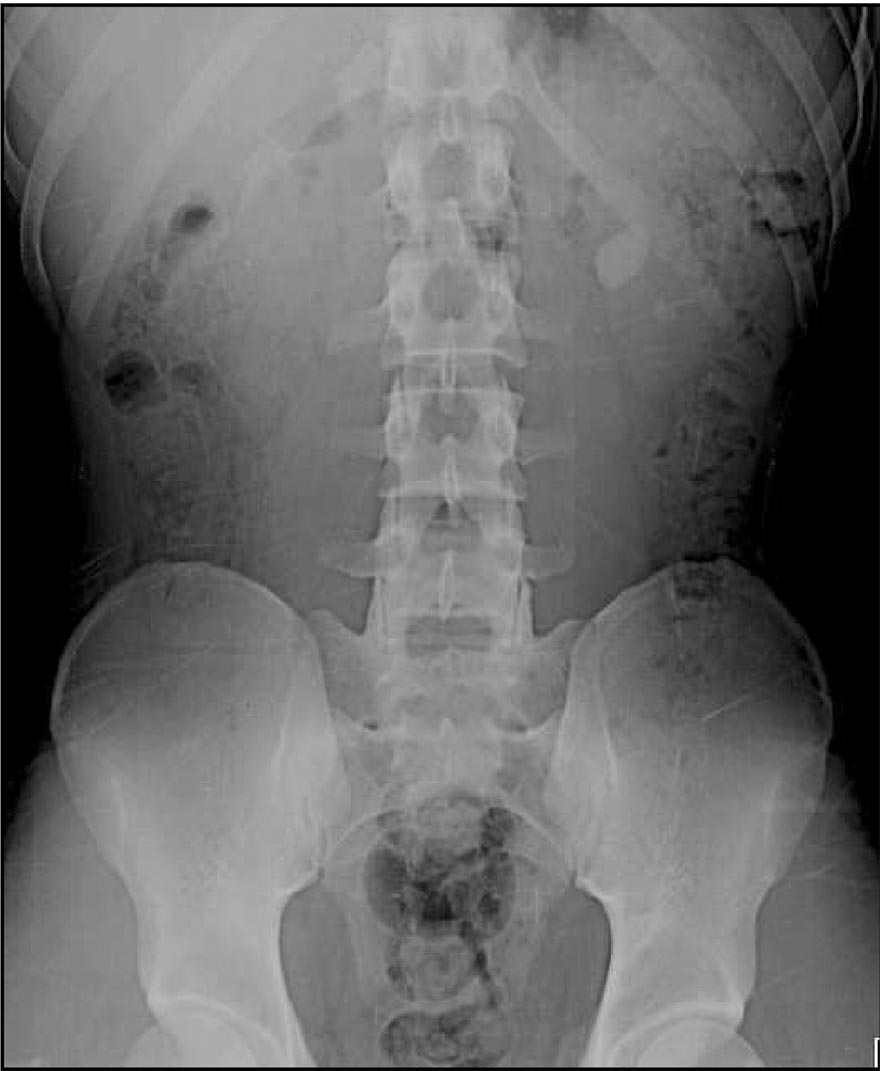
Fig. 5. Imagen radiológica de una litiasis cistínica.
Los cálculos son radiopacos.
La infección urinaria, la hipertensión arterial y la insuficiencia renal puede ser las primeras causas por la que los pacientes cistinúricos requieren atención médica (55). Un reciente estudio concluye que los pacientes cistinúricos presentan mayores concentraciones de creatinina sérica que los formadores de cálculos de oxalato cálcico y que su riesgo de pérdida renal es mucho mayor (56).
La frecuencia de formación de cálculos oscila individualmente entre unas pocas veces a lo largo de la vida hasta varias por año. Como todas las litiasis, las posibles complicaciones incluyen las de la obstrucción del tracto urinario, siendo el fallo renal y la necesidad de hemodiálisis, raros.
Los cálculos de cistina son radiopacos, al menos parcialmente, debido a la densidad del puente disulfuro de la molécula, aunque menos densos que los cálculos de oxalato cálcico. Ocasionalmente son observados por ecografía o TAC helicoidal, que ofrecen la ventaja de demostrar hidronefrosis si se presenta. La tradicional pielografía todavía es útil para demostrar la anatomía del tracto urinario (46).
La profilaxis en el paciente cistinúrico se basará en una abundante hidratación (como mínimo 4 litros de agua al día) con el objeto de mantener concentraciones urinarias de cistina inferiores a 1 mM (concentración de sobresaturación de la cistina) y una densidad urinaria inferior a 1010 g/L (57). Los programas médicos basados en alta diuresis y alcalinización, como primera línea profiláctica y la adición farmacológica de tíoles como segunda evitan la necesidad de procedimientos urológicos a la mitad de los pacientes. Sin embargo, los pacientes que no cumplen con la hiperhidratación se sitúan en un alto riesgo de litogénesis recidivante; siendo el mantenimiento de una diuresis superior a 3 L esencial para la terapéutica, incluso si se está administrando fármacos tío-derivados (58). En cualquier caso, al tratarse de una enfermedad crónica con tratamiento de por vida, el grado de cumplimiento terapéutico del paciente es bajo y precisa de la vigilancia facultativa periódica, especialmente en los litógenos recidivantes.
Se ha preconizado una dieta baja en metionina (aminoácido esencial precursor de la cisteína y cistina que se encuentra en las dietas protéicas de origen animal como carne, pescado y huevos así como en soja, trigo y cacahuetes), aunque su eficacia preventiva no ha podido ser demostrada convincentemente (la cistinuria no desciende en pacientes vegetarianos, que por otra parte aumentan la oxalaturia).
Se aconseja la alcalinización urinaria, puesto que la cistina es más soluble en medio básico, con citrato potásico comenzando con una dosis inicial de 60-80 mEq/día dividida en cuatro dosis (59) y/o dieta rica en zumos cítricos. Los pacientes deben ser instruidos para controlar el pH urinario para mantenerlo alcalino procurando no sobrepasar el de 8 puesto que favorece la precipitación de fosfatos. El bicarbonato de sodio no es aconsejable por su carga sódica, ya que algunos autores recomiendan limitar la ingesta de sodio a menos de 2 g por día (60).
También se han utilizado para la profilaxis y tratamiento médico fármacos derivados del tiol, como: D-penicilamina (Fig. 1), α-mercaptopropionilglicina (α-MPG, tiopronina), captopril, o la N-acetil-cisteína. Que actúan, en el caso de los tres primeros rompiendo, por reducción, el enlace disulfuro de la cistina y formando compuestos mucho más solubles, o, en el caso de la N-acetil-cisteína, por competición con la cistina a nivel tubular. Sin embargo, los considerables efectos secundarios (como reacciones alérgicas, trastornos digestivos, agranulocitosis, proteinuria y síndrome nefrótico) de la administración de estos fármacos disuaden su uso.
La D-penicilamina, sin duda el fármaco mejor estudiado, crea complejos que son 50 veces más solubles que la cistina, su efecto es dosis-dependiente, presenta una gran variedad de efectos secundarios a los que hay que añadir que puede ocasionar deficiencia en vitamina B6 (piridoxina).
Diferentes estudios han demostrado que la α-MPG presenta menores efectos secundarios, puede actuar directamente afectando a la excreción tubular renal de cistina y es más eficaz que la D-penicilamina. La dosis inicial recomendada es de 400 mg/día en cuatro tomas, y un incremento gradual hasta alcanzar la dosis de 800-1200 mg/día en cuatro tomas (60). Además, puede prescribirse a bajas dosis de forma continuada disminuyendo los efectos secundarios (61).
El captopril (un inhibidor de la enzima convertidora de la angiotensia) forma complejos con la cistina 200 veces más solubles, aunque no todos los pacientes son respondedores, parece ser el fármaco de elección en los pacientes en los que ha fracasado la terapéutica médica tradicional y en aquellos que presentan hipertensión concomitante (60).
La excreción urinaria de cistina presenta variaciones intra e interdiarias, por lo que la determinación de la cistinuria en días diferentes y en orina nocturna (en vez de la de 24 h) se ha sugerido para observar los tiempos de sobresaturación de la cistinuria (que pasan desapercibidos en orinas de 24 h) y el subsiguiente ajuste de la terapéutica profiláctica de los pacientes (62).
Se han utilizado soluciones de D-penicilamina o N-acetil-cisteína como disolventes de los cálculos, mediante irrigación con catéteres transureterales o sondas de urostomía.Sin embargo, la quimiodisolución es un procedimiento caro y lento y sin utilidad cuando los cálculos son mixtos conteniendo compuestos cálcicos.
El tratamiento invasivo es el clásico de las litiasis: cirugía abierta, nefrolitotomía percutánea (que también permite el uso de la litotricia ultrasónica o litoclastia con láser-holmio), citoscopía, endourología ureteral con litofragamentación directa con láser holmio, etc. En casos extremos, la combinación de infección y obstrucción puede conducir a una nefrectomía, en este caso procederá el trasplante renal ya que el nuevo riñón injertado no reproducirá el defecto en el transporte tubular de aminoácidos.
La LEOC es de elección para cálculos de diámetro inferior a 1.5 cm, aunque a veces no es eficaz debido a la extrema dureza de los cálculos de cistina y a su naturaleza orgánica que ofrece una débil interfase acústica lo que impide una fácil fragmentación. Esta técnica requiere, para la litiasis cistínica un gran número de ondas de choque y de más alto poder que las otras litiasis. Es importante realizar revisiones periódicas a los enfermos cistinúricos para tratarlos con LEOC cuando los cálculos son iniciales y, por tanto, más fáciles de fragmentar y eliminar. Los cálculos superiores en tamaño a 1,5 cm serán tratados preferentemente por litotricia intracorpórea por aproximación ya anterior o bien retrógada.
Una aproximación inicial al manejo del paciente cistinúrico requerirá un tratamiento preventivo con medidas como hidratación, moderada ingesta de sal (se aconseja una toma diaria inferior a 2 g), y alcalinización oral. La progresión de la enfermedad, hará procedente el tratamiento médico con los fármacos anteriormente citados; sin embargo muchos de los pacientes cistinúricos, y a pesar de las medidas profilácticas, desarrollarán litiasis recidivantes, de las cuales un porcentaje elevado precisarán técnicas urológicas mínimamente invasivas (56,63). Una correcta tipificación del fenotipo/genotipo ayudará a instaurar un tratamiento más adecuado según el riesgo litógeno optando por técnicas mínimamente invasivas para aquellos altamente recidivantes (Tabla I) (19,60).
AGRADECIMIENTO
Al Prof. Dr.Félix Grases Freixedas (Laboratori d´Investigació en Litiasi Renal, Universitat de les lIles Balears) por su inestimable colaboración.
Bibliografía
1. Wollaston WH. On cystic oxide, a new species of urinary calculus. Phil Trans Roy Soc Lond Sci 1810; 100: 223-230. [ Links ]
2. Marcet A. An essay on the chemical history and medical treatment of calculus disorders. En: Longman, Hurst, Rees, Orme y Brown editores. London, 1817. p. 79-88. [ Links ]
3. Noehden GH. Scientific notices –chemistry,cystic oxide- communicated in a letter from Dr. Noehden to Mr. Childre. Annals of Philosophy 1824; 7: 146. [ Links ]
4. Berzelius JJ. Calculus urinaries.Traité Chemistrie 1833; 7: 424-428. [ Links ]
5. Prout W. Traité de la gravelle. París;1823.p. 278. [ Links ]
6. Toel F. Beobachtungen uber Cystinebildung. Ann Chem Pharm 1855; 96:247. [ Links ]
7. Garrod AE. The Croonian lectures on inborn errors of metabolism. Lancet 1908; 2: 1-7, 73-79,142-148 y 214-220. [ Links ]
8. Von Udransky L, Baumann E. Uber das vorkommen von Diaminem, sogenannten Ptomaienen, bei Cystinuria. Z. Physiol Chem 1899; 13: 562. [ Links ]
9. Yeh HL, Frankl W, Dunn MS, Parker P, Hughes B, Gyorgy P. The urinary excretion of amino acids by a cystinuric subject. Am J Med Sci 1947; 214: 507. [ Links ]
10. Stein WH. Excretion of amino acids in cystinuria. Proc Soc Exp Biol Med 1951; 78: 705. [ Links ]
11. Dent CE, Rose GA. Amino acid metabolism in cystinuria. Q J Med 1951; 20: 205-220. [ Links ]
12. Milne MD, Asatoor AM, Edwards KDG, Louhridge LW. The intestinal absorption defect in cystinuria. Gut 1961; 2: 323. [ Links ]
13. Harris H, Mittwoch U, Robson EB, Warren FL. Phenotypes and genotypes in cystinuria. Ann Hum Genet 1955; 20: 57-91. [ Links ]
14. Palacín M, Goodyer P, Nunes V, Gasparini P. Cystinuria. En: Scriver CR, Beaudet AL, Sly WS, Valle D, editores. The metabolic basis of inherited disease. 8ª ed. New York: McGraw-Hill; 2001; Vol III: p. 4909-4932. [ Links ]
15. Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med 2002; 347: 111-121. [ Links ]
16. Goldfarb DS. Cystinuria. Encyclopedia of Life Sciences 2001. Disponible en : www.els.net. [ Links ]
17. Guillén M, Corella D, Cabello ML, García AM, Hernández-Yago J. Reference values of urinary excretion of cystine and dibasic aminoacids classification of patients with cystinuria in the Valencian Community, Spain. Clin Biochem 1999; 32: 25-30. [ Links ]
18. Rosenberg LE, Downing S, Durant JL, Segal L. Cystinuria: biochemical evidence for three genetically distinct diseases. J Clin Invest 1966; 45: 365-371. [ Links ]
19. Dello Strologo L, Pras E, Pontesilli C, Beccia E, Ricci-Barbini V. de Sanctis L, et al. Comparison between SLC3A1 and SLC7A9 cystinuria patients and carriers: a need for a new classification. J Am Soc Nephrol 2002; 13: 2547-2553. [ Links ]
20. Pras E. Cystinuria at the turn of the millenium: clinical aspects and new molecular developments. Mol Urol 2000; 4:409-14. [ Links ]
21. de Sanctis L, Bonetti G, Bruno M, De Luca F, Bisceglia L, Palacín M et al. Cystinuria phenotyping by oral lysine and arginine loading. Clin Nephrol 2001; 56: 467-474. [ Links ]
22. Kanai Y, Endou. Heterodimeric amino acid transporters: molecular biology and pathological and pharmacological relevance. Cur Drug Metab 2001; 4: 339-354. [ Links ]
23. Broer S, Wagner CA. Structure-function relationships of heterodimeric amino acid transporters. Cell Biochem Biophys 2002; 36: 155-168. [ Links ]
24. Fernández E, Carrascal M, Rousaud F, Abian J, Zorzano A, Palacín M, Chillarón J. rBAT-b(0,+) AT heterodimer is the main apical reabsortion system for cystine in the kidney. Am J Physiol Renal Physiol 2002; 283:F540-548. [ Links ]
25. Palacín M. A new family of proteins (rBAT and 4F2hc) involved in cationic and zwitterionic amino acid transport: a tale of two proteins in search of a transport function. J Exp Biol 1994; 196: 123-137. [ Links ]
26. Palacin M, Borsani G, Sebastio G. The molecular bases of cystinuria and lysinuric protein intolerance. Curr Op Genet Develop 2001; 11: 328-335. [ Links ]
27. Goodyer PR, Clow C, Reade T, Girardin C. Prospective analysis and classification of patients with cystinuria identified in a newborn screening program. J Pediatr 1993; 122: 568-572. [ Links ]
28. Lee W, Wells RG, Sabbag RV, Mohandas TK, Hediger MA. Cloning and chromosomal localization of a human kidney cDNA involved in cystine, dibasic, and neutral amino acid transport. J Clin Invest 1993; 91: 1959-1963. [ Links ]
29. Zhang XX, Rozen R, Hediger MA, Goodyer P, Eydoux P. Assignment of the gene for cystinuria (SLC3A1) to human chromosome 2p21 by fluorescence in situ hybridization. Genomics 1994; 24: 413-414. [ Links ]
30. Yan N, Mosckovitz R, Gerber LD, Mathew S, Murty VVS, Tate SS, Udenfriend, S. Characterization of the promoter region of the gene for the rat neutral and basic aminoacid transporter and chromosomal localization of the human gene. Proc Natl Acad Sci 1994; 91: 7548-7552. [ Links ]
31. Purroy J, Bisceglia L, Calonge MJ et al. Genomic structure and organization of the human rBAT gene (SLC3A1). Genomics 1996; 37: 249-252. [ Links ]
32. Pras E, Sood R, Raben N, Aksentijevitch I, Chen XY, Kastner DL. Genomic organization of SLC3A1, a transporter gene mutated in cystinuria. Genomics 1996; 36: 163-167. [ Links ]
33. Ishihara M, Ogura T, Akakura K, Egoshi KI, Mikami K, Nakaya H, Ito H. Cystine transport activity of heterozygous rBAT mutants expressed in Xenopus oocytes. Nephron 2002; 91:276-80. [ Links ]
34. Guillén M, Corella D, Cabello ML, García AM, Portolés O, Hernández-Yago J. Association between M467T and 114C_A variants within the SLC3A1 gene and some pheotypical traits in cystinuria patients from Spain. Hum Genet 2000; 106: 314-320. [ Links ]
35. Chillarón J, Estévez R, Samarzija I, Waldegger S, Testar X, Lang F, Zorzano A, Busch A, Palacin M. An intracellular trafficking defect in Type I cystinura rBAT mutants M467T and M467K. J Biol Chem 1997; 272: 9543-9549. [ Links ]
36. Goodyer P, Saadi I, Ong P, Elkas G, Rozen R. Cystinuria subtype and the risk of nephrolithiasis. Kidney Int 1998; 54: 56-61. [ Links ]
37. Bisceglia L., Calonge MJ, Totaro A et al. Localization, by linkage analysis, of the cystinuria type III gene to chromosome 19q13.1. Am J Hum Genet 1997; 60: 611-616. [ Links ]
38. Feliubadalo L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, et al. International Cystinuria Consortium. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (b(0,+)AT) of rBAT. Nature Genet 1999; 23: 52-57. [ Links ]
39. Font MA, Feliubadalo L, Estivill X, Nunes V, Golomb E, Kreiss Y, et al. International Cystinuria Consortium. Functional analysis of mutations in SLC7A9, and genotype-phenotype correlation in non-Type I cystinuria. Hum Molec Genet 2001; 10: 305-316. [ Links ]
40. Brand E, Harris MM, Biloon S. Cystinuria: Excretion of a cystine complex which descomposes in the urine with the liberation of free cystine. J Biol Chem 1930; 86: 315-359. [ Links ]
41. Finocchiaro R, D’Eufemia P, Celli M, Zaccagnini M, Viozzi, Troiani P et al. Uselfulness of cyanide-nitropruside test in detecting incomplete recessive heterozygotes for cystinuria: a standardized dilution procedure. Urol Res 1998; 26:401-405. [ Links ]
42. Pinto B. Cistinuria. En: Pinto B. Litiasis renal. 2ª ed. Barcelona: Masson-Salvat; 1993; p.135-43. [ Links ]
43. Grases F, Villacampa AI, Costa-Bauzá A, Otakar Söhnel. Uric acid calculi: types, etiology and mechanisms of formation. Clin Chim Acta 2000; 302:89-104. [ Links ]
44. Vilanova MA. Métodos habituales. En: Pinto B. Litiasis renal. 2ª ed. Barcelona: Masson-Salvat; 1993; 12-13. [ Links ]
45. Giugliani R, Ferrari I, Greene LJ. An evaluation of four methods for the detection of heterozygous cystinuria. Clin Chim Acta 1987; 164:227-233. [ Links ]
46. Soupart P. Free amino acids of blood and urine in the human. En: Amino acid pools. Ed. Holden JT. Amsterdam-Elsevier 1962; 220-262. [ Links ]
47. Guillén M, Corella D, Cabello ML, González JI, Hernández-Yago J. Manifestaciones fenotípicas de la cistinuria: estudio de 20 familias en la Comunidad Valenciana. Med Clin (Barc) 2000; 115: 610-616. [ Links ]
48. Watanabe H, Sugahara K, Inoue K, Fujita Y, Kodama H. Liquid chromatography-mass spectronic analysis for screening of patients with cystinuria, and identification of cystine stone. J Chromatogr 1991; 568: 445-450. [ Links ]
49. Guillén M, Corella D, Cabello ML, García AM, Sáiz C, González JI, Hernández-Yago J. Estudio genético y molecular de la cistinuria en la Comunidad Valenciana. Med Clin (Barc) 1999; 113: 321-326. [ Links ]
50. Guillén M, Corella D, Cabello ML, Saiz C, Hernández-Yago J. Sensibilidad, especificidad y valor predictivo del análisis genético de variantes en el gen SLC3A1 aplicado al diagnóstico de cistinuria en la población española. Rev Clin Esp 2001; 201:256-259. [ Links ]
51. Pras E, Kreiss Y, Frishberg Y, Aksentijevich I, Kastner DL. Refined mapping of the CSNU3 gene to a 1.8-Mb region on chromosome 19q13.1 using historical recombinants in Libyan Jewish cystinuria patients. Genomics 1999; 60:248-50. [ Links ]
52. Levy FL, Adams-Huet B, Pak CY. Ambulatory evaluation of nephrolitiasis: an update of 1980 protocol. Am J Med 1995; 98: 50-59. [ Links ]
53. Asatoor AM, Crouchman MR, Harrison AR, Light FW; Loughridge LW, Milne MD et al. Intestinal absortion of oligopeptides in cystinuria. Clin Sci 1971; 41: 23-33. [ Links ]
54. Scriver CR, Whelan DT, Clow CL, Dallaire L. Cystinuria: increased prevalence in patients with mental disease. New Eng J Med; 1970; 283: 783-786. [ Links ]
55. Rodríguez Fernández LM, Lapeña López de Armentia S. Cistinuria. Revisión teórica. Bol Pediatr 1997; 37: 73-77. [ Links ]
56. Assimos DG, Leslie SW, Ng C, Streem SB, Hart LJ. The impact of cystinuria on renal function. J Urol 2002; 168:27-30. [ Links ]
57. Rutchik SD, Resnick MI. Cystine calculi: diagnosis and management. Urol Clin North Am 1997; 24: 163-171. [ Links ] 5
58. Barbey F, Joly D, Rieu P, Mejean A, Daudon M, Jungers P. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol 2000; 163: 1419-1423. [ Links ]
59. Fjellstedt E, Denneberg T, Jeppsson JO, Tiselius HG. A comparison of the effects of potassium citrate and sodium bicarbonate nin the alkalinization of urine in homozygous cystinuria. Urol Res 2001; 29:295-302. [ Links ]
60. Ng CS, Streem SB. Medical and surgical therapy of the cystine stone patient. Curr Opin Urol 2001; 4: 353-358. [ Links ]
61. Berio A, Piazzi A. Prophylaxia of cystine calculosis by alpha-mercaptopropionyl-glycine administered continuously or every other day. Boll Soc Ital Biol Sper 2001; 77:34-41. [ Links ]
62. Fjellstedt E, Denneberg T, Jeppsson JO, Christensson A, Tiselius HG. Cystine analyses of separate day and night urine as a abasis for the management of patients with homozygous cystinuria. Urol Res 2001; 29:303-10. [ Links ]
63. Rousaud F, Gracia S, Palacín M, Nunes V, Millán F, Oliver A, Rousaud A. Cistinuria y litiasis renal de cistina. Estudio y enfoque terapéutico. Arch Esp de Urol 2001; 54: 989-996. [ Links ]

