










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.22 no.1 ene. 2005
| Glomerulonefritis fibrilar e inmunotactoide: descripción de un caso y revisión de la literatura F. J. VERA MÉNDEZ, M. MOLINA NÚÑEZ1, M. A. HERNÁNDEZ GARCÍA1, J. GARCÍA SOLANO2 Servicios de Medicina Interna, 1Nefrología y 2Anatomía Patológica. Hospital Santa María del Rosell. Cartagena. Murcia
|
| FIBRILLARY AND IMMUNOTACTOID GLOMERULONEPHRITIS: REPORT OF A CASE AND REVIEW OF THE LITERATURE
| ||
| RESUMEN Describimos el caso de una mujer de 67 años de edad que presentó síndrome nefrótico e insuficiencia renal rápidamente progresiva. La nefropatía se caracterizó por la evidencia a través de microscopía electrónica de depósitos de fibrillas orientadas al azar con diámetro aproximado de 18-20 nm. La inmunofluorescencia fue negativa tanto para inmunoglobulinas como complemento. La paciente fue diagnosticada de una atípica glomerulopatía fibrilar con ausencia de depósitos inmunes y evolucionó rápidamente a enfermedad renal terminal poco tiempo después. Revisamos en la literatura nuevos aspectos clínicos y patogenéticos relacionados con la glomerulopatía fibrilar e inmunotactoide. PALABRAS CLAVE: Glomerulonefritis fibrilar inmunotactoide. | ABSTRACT We describe the case of a 67 year -old female with nephrotic syndrome and rapidly progressive renal failure. The nephropaty was characterized by deposits of randomly oriented fibrils with a diameter of about 18-20 nm on electron microscopy. Immunofluorescence microscopy was performed and there was no staining for immunoglobulins and complement. We diagnosed atypical fibrillary glomerulopathy with absence of immune deposits. The patient developed end-stage renal failure rapidly. We review in the literature new clinical and pathogenetic features related to fibrillary and immunotactoid glomerulopathy. KEY WORDS: Glomerulonephritis fibrillary immunotactoid. |
Vera Méndez FJ, Molina Núñez M, Hernández García MA, García Solano J. Glomerulonefritis fibrilar e inmunotactoide: descripción de un caso y revisión de la literatura. An Med Interna (Madrid) 2005; 22: 35-38.
Trabajo aceptado: 1 de septiembre de 2004
Correspondencia: Francisco Jesús Vera Méndez. C/Tierno Galván, 16-6ºA, 30203. Cartagena. Murcia. e-mail: fco-vera@ono.com
INTRODUCCIÓN
Esta glomerulopatía fue descrita por vez primera en 1977, cuando Rosenmann y Eliakim observaron a nivel del glomérulo, material fibrilar similar al amiloide pero que no compartía las características congofílicas de esta última. Más tarde, Duffy y cols describieron en 1983 a 8 pacientes con hipertensión, hematuria y proteinuria en cuyas biopsias se objetivaron depósitos de fibrillas dispuestas al azar en torno a 20 nm a nivel mesangial y transmembranoso, utilizando el término de fibrilar para designar la ultraestructura de dichos depósitos.
Sin embargo, el término de glomerulonefritis fibrilar (GF) se acuñó en 1987 por Alpers y cols., al describir en 7 casos la existencia a nivel extracelular de depósitos constituidos por fibrillas orientadas al azar y de un diámetro superior al amiloide (10-20 nm), las cuales contenían inmunoglobulinas y complemento.
De forma casi paralela, en 1980 Schwartz y Lewis describen otra glomerulopatía caracterizada por la existencia de estructuras microtubulares de mayor tamaño (aproximada-mente 27 nm), con depósitos inmunes monoclonales IgG kappa y con una especial disposición en bandas paralelas, recibiendo el nombre esta entidad de glomerulopatía inmunotactoide (GIT).
Hoy en día sigue existiendo confusión a la hora de utilizar una terminología que defina mejor a estas glomerulopatías. Pese a la divulgación durante años por varios autores del término "glomerulonefritis fibrilar inmunotactoide", aludiendo a que ambas glomerulopatías formarían una misma entidad clínico-patológica sin criterios clínicos y patogénicos suficientes como para justificar una subclasificación, son cada vez más numerosas las opiniones de autores que intentan separar a ambas entidades en virtud no sólo de las diferencias ultraestructurales, sino a la tipificación del tipo de depósito inmune y a las cada vez más importantes implicaciones clínicas y renales que se documentan en las series y casos publicados.
Mujer de 67 años, con historia de HTA esencial conocida y tratada durante cinco años con IECA y tiazidas, hipotiroidismo primario bien controlado en tratamiento sustitutivo con L-tiroxina y antecedente de poliartrosis generalizada con consumo ocasional de paracetamol y/o AINE. Acude al Servicio de Urgencias en mayo de 2002 tras presentar clínica de edemas progresivos en miembros inferiores, los cuales se hacen generalizados además de disnea de esfuerzo progresiva en los cinco meses previos al ingreso. En planta se constataron signos de anasarca. En cuello se observó ligera IY a 45º, destacando en la auscultación cardiopulmonar soplo protosistólico ligero en foco aórtico e hipofonesis en ambas bases pulmonares.
De entre los parámetros analíticos, inmunológicos y serológicos destacaron: Hb 10,36, Hto 32,29, VCM 78, HCM 25,52, 6.700 LT (N58,68%, L 27,92%, M 6,30%, E 5,84%, B5,84%), 257.000 Pl, fibrinógeno 785; morfología en sangre periférica: anisocitosis con anisocromía, anisotrombocitosis con presencia de algunas plaquetas grandes parcialmente degranuladas y normalidad en serie blanca; Cr 1,9 mg/dl, urea 61 mg/dl, aclaramiento Cr 25 ml/min, Col T 213 mg/dl, TG 275 mg/dl, Prot totales 4,0 g/dl, albúmina 1,7 g/dl, IgG 495 UI/l, LDH 556 UI/dl, hierro sérico 40 mcg/dl, antitrombina III 57%; proteinograma: Alb 39%, alfa 1 globulina 9,5%, alfa 2 globulina 25%, beta globulina 13%, gammaglobulina 11,9%, CA 125 66,39 UI/ml siendo GPT, GOT, FA GGT, Ac úrico, BT y BD, CK, calcio iónico, fósforo, ferritina, Ig M, IgA, T4 Libre, TSH, B2 microglobulina, PCR, FR, PTH, CEA, CA 19,9, CA 15,3, alfa fetoproteína, C3, C4 y cadenas ligeras en sangre y orina dentro de la normalidad.
La determinación de ANA screening, antiDNA y ENA por Elisa fueron negativas. La determinación de ANCA (IFI) , Ac antitiroideos (antitiroglobulina y antiperoxidasa), crioglobulinas así como serología para virus VIH, VHC y VHB fueron negativos. En el estudio bioquímico de orina destacó un PH 6, densidad 1.030, con 250 eri/mcl y 500 mg proteínas/dl en anormales, detectándose abundantes cilindros hialino-granulosos y escasos céreos y celulares en el sedimento. El análisis de orina de 24 horas mostró una proteinuria en rango nefrótico (microproteinuria 11.898 mg/dl, microalbuminuria 7548,8 mg/dl).
En la radiografía de tórax se observó derrame pleural bilateral de predominio izquierdo y signos de redistribución vascular. La Rx abdomen mostró colelitiasis sin otros datos de interés, y en el ECG destacó un ritmo sinusal con bajos potenciales eléctricos en derivaciones bipolares con aplanamiento de onda T. Se realizó ecocardiograma en el que se objetivó una hipertrofia ventricular izquierda ligera y de predominio septal (15 mm), siendo el tamaño del VI y la AI normales así como la función sistólica global y segmentaria. En el estudio doppler se descartó patología restrictiva (onda A>E).
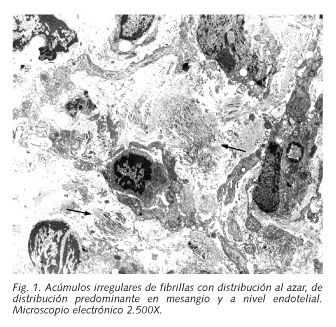
La ecografía abdomino-renal informó de colelitiasis no complicada con presencia de riñones normales en tamaño, morfología y ecogenicidad. En la descripción microscópica de la biopsia renal se observó intenso depósito eosinófilo pálido a nivel mesangial y en la pared capilar, sin aumento de celularidad mesangial ni presencia de células inflamatorias en los 16 glomérulos examinados. Dicho depósito fue homogéneo por todo el ovillo, siendo PAS positivo y negativo para metenamina de plata. En el estudio de inmunofluorescencia no se observaron depósitos de inmunoglobulinas ni de C3. Con rojo Congo no se observó tinción positiva ni se obtuvo birrefringencia verdosa con luz polarizada. Ante estos hallazgos compatibles con enfermedad de depósito renal no amiloidea, se realizó una 2ª biopsia renal para obtención de muestra y posterior análisis por microscopía electrónica, en donde se identificó las características ultraestructurales del depósito como material fibrilar difusamente distribuido predominantemente por el mesangio y a nivel endotelial, en donde los depósitos protruían sobre el citoplasma de las células endoteliales.
Dicho material fibrilar se disponía en acúmulos irregulares con diversa orientación, lo que también ocurría con las fibrillas en el interior de cada uno de los acúmulos (Fig. 1), con un tamaño de 18-20 nm (Fig. 2), siendo todo ello compatible con glomerulopatía fibrilar.

Durante el seguimiento ambulatorio, la paciente presentó rápido deterioro de la función renal (Cr 5,3 mg/dl, urea 90 mg/dl), descartándose patología obstructiva por lo que la paciente entró en programa de hemodiálisis a finales del mes de junio de 2002. La paciente falleció en febrero de 2003 por causas extrarrenales.
DISCUSIÓN
La glomerulonefritis fibrilar (GF) e inmunotactoide (GIT) forman parte de las denominadas enfermedades renales de depósito no amiloideas, entidades éstas poco frecuentes (1% y 0,2% respectivamente del total de biopsias renales de importantes series) (1-3) y de reciente descripción en la literatura. Típicamente ofrecen al microscopio óptico un aspecto similar al de la amiloidosis, con existencia de expansión del mesangio y engrosamiento de la pared capilar, pero a diferencia de ésta, casi nunca se ve efectado el intersticio, los vasos y los túbulos, además de encontrar negatividad para amiloide con Rojo Congo y Tioflavina T. Los depósitos son PAS positivos y metenamina de plata negativos.
En la GF, el patrón de inmunofluorescencia corresponde a la distribución de los depósitos observados ultraestructuralmente En más del 95% de las muestras en las series publicadas son positivas para IgG y C3, 60% para IgM y 30% para IgA (4). Ambas cadenas kappa y lambda son positivas en la mayoría de los pacientes (1,2). Sólo en un 10% de los datos ofrecidos en la literatura se describen depósitos monoclonales, por lo general IgG Kappa. Iskandar y cols1 en una amplia serie publicada en 1992 sobre GF (excluyendo los casos de GIT), observó en el estudio de subclases, que la mayoría de los depósitos estaban conformados por IgG4 policlonal de forma dominante o exclusiva, planteando la hipótesis de la necesidad de una relativa homogeneidad de la Ig en los depósitos inmunes para favorecer la formación fibrilar. En la GIT, también los depósitos más frecuentes han sido de IgG y C3, sin embargo los depósitos de IgG4 policlonal suelen ser menos frecuentes y por contra la monoclonalidad de los depósitos parece ser la nota dominante a diferencia de lo que ocurre en la GF.
Por tanto los hallazgos inmunohistoquímicos indican que los depósitos de microfibrillas estarían conformados por inmunoglobulinas y complemento, como demostraron Yang y cols. (5) en microscopía electrónica, sugiriendo éstos que las fibrillas representarían componentes proteicos polimerizados. Korbet y cols. (6) propusieron igualmente la necesidad de uniformidad de la inmunoglobulina en los depósitos para constituir la base de las estructuras fibrilares.
Sin embargo, en casos muy excepcionales como los descritos por Churg (7) (dos casos de GF) y Vigil y cols. (8) (1 caso de GIT asociado a mieloma múltiple) y de forma similar a la paciente que describimos en nuestro artículo, se describe ausencia de inmunoglobulinas y complemento al aplicar tecnicas inmunohistoquímicas, lo cual revelaría la gran heterogeneidad de esta entidad y la necesidad de incluir una variedad de precursores en el que figuren no solo las inmunoglobulinas sino otras sustancias aún no identificadas. Reciente es la propuesta realizada por Rostagmo y cols. (9) sobre la necesidad de la existencia de un precursor sérico como origen de los depósitos fibrilares, sugiriendo el importante rol que ejercerían los complejos conformados por inmunoglobulina-fibronectina en la patogénesis de la GF.
El estudio de microscopía electrónica se hace imprescindible para el diagnóstico ultraestructural. La variante fibrilar, se caracteriza por la existencia de pequeñas fibrillas dispuestas al azar con un tamaño que oscila en torno a 12-20 nm (1,2) y que por definición son de mayor tamaño que las del amiloide (6-10 nm), de aspecto sólido en los aumentos de rutina aunque en grandes aumentos algunos autores han identificado la existencia de luces (4). En la variante inmunotactoide , las microfibrillas son mas grandes (en torno a 30 nm), suelen tener luces visibles conformando microtúbulos y característicamente se agrupan formando bandas paralelas (10). Típicamente los depósitos fibrilares concuerdan con la distribución de los depósitos inmunes, localizándose con más frecuencia a nivel del mesangio y también en las paredes capilares distribuidas en los espacios subendotelial y subepitelial así como a través de la membrana basal. Los depósitos están usualmente confinados al glomérulo aunque en raras ocasiones se pueden observar en el intersticio rodeando a los capilares peritubulares. Pese a la consideración de estas enfermedades de depósito como primarias o exclusivas a nivel renal, se ha identificado un caso aislado de GF con afectación pulmonar (11), manifestándose en forma de hemorragia alveolar y demostrándose en la autopsia microfibrillas de morfología y tamaño similares a los hallados en el glomérulo. De igual manera se ha descrito varios casos de GIT con depósitos fibrilares extrarrenales a nivel hepático y médula ósea asociados a gammapatía monoclonal (12,13).
La forma de presentación clínica de ambas glomerulopatías son prácticamente idénticas; las dos se acompañan en la mayoría de los casos de proteinuria elevada, por lo general en rango nefrótico junto a hematuria e hipertensión y hasta en la mitad de los casos con insuficencia renal (14). Los autores partidarios de la diferenciación de ambas entidades se apoyan no sólo en las diferencias ultraestructurales, sino en la trascendencia clínica que ello conllevaría. De un lado, Alpers (10) propone considerar la GIT como una entidad distinta a la GF por su fuerte asociación a procesos lifoproliferativos y disproteinemias a diferencia de lo que ocurre con esta última.
En 1993, Fogo y cols. (2) en una serie constituida por 26 casos de GF y 6 casos de GIT observaron diferencias clínicas notables. En este trabajo se objetivó que los pacientes con GF presentaban un peor pronóstico renal, describiéndose en sólo 1 caso mieloma múltiple. Por el contrario en 4 de los 6 pacientes con GIT se evidenció asociación con paraproteinemia, presentaron una edad media más avanzada, y se comprobó a largo plazo un mejor pronóstico renal. En otra gran serie reciente presentada por Bridoux y cols. (15), se incluyeron 14 casos de GIT, en 10 de los cuales se identificaron procesos neoplásicos hematológicos. Por otra parte, en ninguno de los 9 casos de GF que se describieron en este trabajo, se observó asociación con procesos linfoproliferativos o discrasias sanguíneas. Aunque estos autores no encontraron en ambas entidades diferencias significativas en cuanto a supervivencia renal, sí se pudo comprobar que la administración de quimioterapia permitió la remisión del síndrome nefrótico en los 10 pacientes con GIT y neoplasia hematológica.
Los partidarios de considerar la glomerulopatía fibrilar e inmunotactoide como una misma entidad clínico patológica (4), se apoyan en que las diferencias ultrestructurales no se corresponden con diferencias clínicas validadas estadísticamente y a la falta de consenso real en diferentes estudios sobre la existencia de una mayor asocición de GIT con enfermedades hematológicas. Otras autores prefieren utilizar el término de "GN fibrilar inmunotactoide" en virtud de los numerosos casos de solapamiento ultraestructural entre ambas variantes y a la inconstante asociación entre monoclonalidad y tamaño de la fibrilla (16). En opinión de Brady y cols. (14), el término de GN fibrilar inmunotactoide definiría mejor esta enfermedad por depósito glomerular hasta que nuevos estudios arrojen más evidencias sobre la necesidad de diferenciar estas dos variantes.
Aunque numerosas asociaciones con otras enfermedades se han descrito como procesos linfoproliferativos (10,15), mieloma (2,8), Sd de Sjögren (2), vasculitis leucocitoclástica (17), crioglobulinemia (18), la mayoría de casos se tratan de idiopáticos (19). En favor de la pluripatogenia de estas entidades merece destacar la comunicación de recientes casos de GF y GIT en pacientes con infección por el virus de la hepatitis C (19,20), informándose en uno de ellos la normalización de la función renal tras completar terapia con interferón (20). También recientemente se han comunicado 2 casos de coinfección VIH y VHC asociados a estas glomerulopatías (21). El tratamiento médico actual de la glomerulonefritis fibrilar e inmunotactoide en los casos en los que no se describe enfermedad sistémica subyacente o asociada, es insatisfactorio con corticoterapia e inmunosupresión (8,15). Se ha ensayado el transplante renal, observándose en la mayoría de los casos recurrencia de la enfermedad aunque el deterioro de la función renal evolucionó invariablemente más lentamente, por lo que se considera una opción atractiva en pacientes con enfermedad renal terminal (22).
En base a lo expuesto, debemos considerar a la glomerulopatía fibrilar e inmunotactoide como enfermedades renales de etiopatogenia aún poco aclaradas y definidas, en las que cabe descartar de forma razonable enfermedades sistémicas (neoplasias hematológicas, enfermedades inflamatorias o procesos infecciosos virales) en las cuales, la aplicación de un tratamiento específico y temprano pudiera modificar la historia natural de las mismas y el de la glomerulopatía. La descripción de ausencia de depósitos inmunes en la inmunofluorescencia en el caso que presentamos, supone un hallazgo excepcional en la glomerulopatía fibrilar e inmunotactoide, lo que refuerza la tesis de la heterogeneidad de la patogenia de la enfermedad, debiendo considerar la existencia de otros precursores séricos aún no identificados (al igual que ocurre con la amiloidosis), como responsables de la conformación de la microfibrilla en el glomérulo renal.
AGRADECIMIENTOS
Agradecer a D. Joaquín Moya Biosca, técnico del Servicio de Anatomía Patológica del Hospital Santa Mª del Rosell y al Profesor del Departamento de Anatomía Patológica de la Facultad de Medicina de la Universidad de Murcia, Dr Vicente Vicente Ortega por su inestimable ayuda y colaboración en la preparación, procesamiento y exámen de las muestras enviadas al Departamento de Microscopía Electrónica.
Bibliografía
1. Iskandar S, Falk R, Jannete C. Cinical and pathology features of fibrillary glomerulonephritis. Kidney Int 1992; 42: 1401-7. [ Links ]
2. Fogo A, Nauman Q, Horn R. Morphologic and clinical features of fibrillary glomerulonephritis versus immunotactoid glomrulophaty. Am J Kidney Dis 1993; 3: 367-77. [ Links ]
3. Korbet SM, Schwartz MM, Rosenberg BF, Sibley RK, Lewis EJ. Immunotactoide glomerulopathy. Medicine 1985; 64: 228. [ Links ]
4. Korbet SM, Schwartz MM, Lewis EJ. The fibrillary glomerulopathies. Am J Kidney Dis 1994; 23: 751-65. [ Links ]
5. Yang GCH, Nieto R, Stachura I, Gallo GR. Ultraestructural immunohistochemical localization of polyclonal IgG, C3 and amyloid P component on the Congo red-negative amyloid-like fibrils of fibrillary glomerulopathy. Am J Pathol 1992; 141: 409-19. [ Links ]
6. Korbet SM, Schwartz MM, Lewis EJ: Immunotactoid glomerulopathy. Am J Kidney Dis 1991; 17: 247-57. [ Links ]
7. Churg J, Venkataseshan S. Fibrillary glomerulonephritis without immunoglobulin deposits in the kidney. Kidney Int 1993; 44: 837-42. [ Links ]
8. Vigil A, Oliet A, Gallar P, Ortega O, Rodríguez Villarreal I, Picazo L, Pinedo F. Rapidly progressive immunotactoid glomerulonephritis and multiple myeloma. Nephron 1998; 79: 238-40. [ Links ]
9. Rostagno A, Vidal R, Kumar A. Fibrillary glomerulonephritis related to serum fibrillar immunoglobulin-fibronectin complexes. Am J Kidney Dis 1996; 28: 676-684. [ Links ]
10. Alpers CE. Immunotactoid (microtubular) glomerulopathy: an entity distict from fibrillary glomerulonephritis? Am J Kidney Dis 1992;19: 185-91. [ Links ]
11. Masson RG, Rennke HG, Gottlieb MN. Pulmonary hemorrhage in a patient with fibrillary glomerulonephritis. N Engl J Med 1992; 326: 36-39. [ Links ]
12. Strom EH, Hurwitz N, Mayr AC, Krause PH, Mihatsch MJ. Immunotactoidlike glomerulopathy with massive fibrillary deposits in liver and bone marrow gammopathy. Am J Nephrol 1996; 16: 52:3 -8. [ Links ]
13. Daas N, Kleinman Y, Polliak A. Glomerupathy with massive bone marrow deposits in a patient wth IgM kappa monoclonal gammopathy. Am J Kidney Dis 2000; 2: 395-399. [ Links ]
14. Brady HR. Fibrillary glomerulopathy. Kidney Int 1998; 53: 1421-29. [ Links ]
15. Bridoux F, Hugue V, Coldefy O, Goujon JM, Bauwens M, Sechet A, et al. Fibrillary glomerulonephritis and immunotactoid (microtubular) glomerulopathy are associated with distinct immunologic features. Kidney Int 2002; 62: 1764-1775. [ Links ]
16. García I, Velenzuela M. Glomerulopatías con depósitos fibrilares. Rev Esp Patol 2002; 35: 167-176. [ Links ]
17. Orfila C. Meeus F, Bernadet P, Lepert JC, Suc JM. Immunotactoid glomerulopathy and cutaneus vasculitis. Am J Nephrol 1991; 11: 67-72. [ Links ]
18. King J, Culppeper M, Corey R, Tucker A, Lajoie G, Howell D. Glomerulopathies with fibrillary deposits. Ultraestruct Pathol 2000; 24: 5-21. [ Links ]
19. Markowitz G, Cheng J, Colvin R, TrebbinW, D'Agati V. Hepatitis C infection is associated with fibrillary glomerulonephritis and immunotactoid glomerulopathy. J Am Soc Nephrol 1998; 9 :2244-52. [ Links ]
20. Coroneos E, Truong L, Oliveo J. Fibrillary glomerulonephritis associated with hepatitis C viral infection. Am J Kidney dis 1997; 29: 132-35. [ Links ]
21. Haas M, Rajaraman S, Ahuja T, Kittaka M, Cavallo T. Fibrillary/ Inmmunotactoid glomerulonephritis in HIV-positive patients: A report of three cases. Nephrol Dial Transplant 2000; 15:1679-1683. [ Links ]
22. Pronovost PH, Brady HR, Gunning ME, Espinoza O, Rennke HG. Clinical features, predictors of disease progression and results of renal transplantation in fibrillary/immunotactoid glomerulopathy. Nephrol Dial Transplant 1996; 11: 837-842. [ Links ]

