










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.23 no.2 feb. 2006
Histiocitosis pulmonar de células de Langerhans
Pulmonary Langerhans' cell histiocytosis
A. M. García Bernárdez, C. C. Álvarez Cuesta, M. E. Rivas Carmenado, E. Vallina Álvarez, J. M. Arribas Castrillo
Servicio de Medicina Interna I. Hospital Universitario Central de Asturias. Oviedo. Asturias
Dirección para correspondencia
RESUMEN
La histiocitosis pulmonar de células de Langerhans forma parte de un espectro de enfermedades caracterizadas por la proliferación monoclonal y la infiltración de distintos órganos por células de Langerhans. Es una enfermedad pulmonar intersticial de etiología desconocida que ocurre casi exclusivamente en pacientes fumadores. El curso de esta enfermedad en adultos es impredecible, oscilando desde formas benignas autolimitadas, hasta formas malignas con evolución progresiva hacia el fallo respiratorio y la muerte.
Presentamos el caso de un paciente diagnosticado de histiocitosis pulmonar de células de Langerhans que presentó una mejoría clínica y radiográfica tras abandonar el hábito tabáquico.
Palabras clave: Histiocitosis. Células de Langerhans. Pulmón.
ABSTRACT
Pulmonary Langerhans'- cell histiocytosis belongs to a spectrum of diseases characterized by monoclonal proliferation and infiltration of organs by Langerhans'cells. It is an uncommon interstitial lung disease of unknown etiology occurring almost exclusively in cigarette smokers. It's course in adults is variable and unpredictable, ranging from benign self-limiting types with spontaneous regression to slowly progressive malignant disease that leads to respiratory failure and death.
We report one patient diagnoses of pulmonary Langerhans'cell histiocytosis who experimented an objetive radiographic improvement and disappearance of symptoms after smoking cessation.
Key words: Histiocytosis. Langerhans cells. Lung.
Introducción
La histiocitosis de células de Langerhans es una enfermedad poco frecuente, caracterizada por el acúmulo y proliferación de histiocitos y eosinófilos, afectando órganos y sistemas, de forma aislada o múltiple.
La forma localizada de histiocitosis de células de Langerhans se denominaba anteriormente granuloma eosinófilo, mientras que las variantes multisistémicas se conocían con los nombres de enfermedad de Letterer-Siwe (forma multifocal, progresiva y crónica) y enfermedad de Hand-Schuller-Christian (forma multisistémica, diseminada y aguda). En 1953, Lichtenstein aplico el término de histiocitosis X al conjunto de enfermedades previamente designadas como granuloma eosinófilo, enfermedad de Letterer-Siwe y enfermedad de Hand-Schuller-Christian (1,2). Actualmente, el Writing Group of the Histiocyte Society recomienda usar el término histiocitosis de células de Langerhans (HCL) sustituyendo al de histiocitosis X.
El término histiocitosis pulmonar de células de Langerhans (HPCL) se usa para hacer referencia a un tipo de histiocitosis que aparece en adultos y que afecta al pulmón de forma aislada o bien en asociación con la afectación de otros órganos. Para evitar confusiones, la Histiocyte Society ha establecido un sistema de clasificación simplificado (3) (Tabla I).

Caso aportado
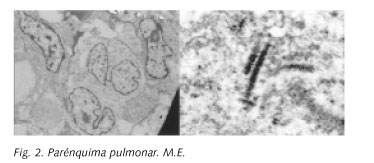
Presentamos el caso de un varón de 29 años de edad, que acude a nuestra consulta por un cuadro de un mes de evolución que consistía en fiebre diaria de 39º-40º, dolor intenso de características pleuríticas en el hemitórax izquierdo y tos con expectoración abundante de color negruzco. No tenía antecedentes médicos ni quirúrgicos de interés. Era fumador de 20 cigarrillos/día desde hacía 10 años. La exploración física fue rigurosamente normal, exceptuando la auscultación pulmonar en la que presentaba una disminución global del murmullo vesicular con zonas aisladas de egofonía. Se realizaron diversos estudios analíticos que incluyeron: hemograma, bioquímica general, gasometría arterial basal, aclaramiento de creatinina, calciuria, proteinograma, dosificación de inmunoglobulinas y complemento, proteína C reactiva, factor reumatoide, determinación de la enzima convertidora de angiotensina y marcadores biológicos, siendo todos los resultados normales. La espirometría realizada mostró un patrón restrictivo. Se realizó un test de difusión pulmonar que puso de manifiesto una difusión disminuida (65%), compatible con una reducción de la superficie de intercambio. En la radiografía simple de tórax se apreciaba un patrón intersticial bilateral. Se realizó una TC torácica (Figs. 1a, 1b) donde se vieron múltiples lesiones quísticas pulmonares, ninguna de ellas mayor de 2 centímetros, distribuidas en ambos pulmones especialmente en los lóbulos superiores y medios. Se observó también una lesión lítica a nivel del cuarto arco costal izquierdo (Fig. 1c). Se solicitó al Servicio de Cirugía Torácica la realización de una biopsia pulmonar abierta. Se tomaron muestras de los lóbulos superior e inferior del pulmón izquierdo. El estudio histopatológico mostró en el parénquima pulmonar, una infiltración por eosinófilos y por células mononucleares con núcleos arriñonados típicos de las células de Langerhans. Las tinciones inmunohistoquímicas S-100 y CD1a fueron positivas. Se realizó estudio de microscopía electrónica y se observaron las características células de Langerhans (Fig. 2a) con núcleo polilobulado y con profundas indentaduras. Se vieron también estructuras formadas por dos dobles membranas apuestas, separadas por una estructura densa que cumple las características de un gránulo de Birbeck en su porción tubular (Fig. 2b). Con el diagnóstico de HPCL se recomendó al paciente abandono del hábito tabáquico y se le citó para realizar TC torácico de control a los 6 meses. En este estudio se observó la desaparición casi completa de las lesiones quísticas persistiendo la imagen lítica costal. (Fig. 1d). Clínicamente el paciente presentó una mejoría importante estando asintomático a no ser por la persistencia de un leve dolor costal en el hemitórax izquierdo que se correspondía con la lesión ósea observada en la TC.

Discusión
Las células de Langerhans son células diferenciadas de la línea de los histiocitos que funcionan como presentadoras de antígenos. Miden aproximadamente 12 micras y tienen un citoplasma abundante y acidófilo con escasas organelas y núcleos de contornos arriñonados. El estudio con microscopía electrónica demuestra la presencia de unas estructuras con forma de raqueta que suelen agruparse cerca del aparato de Golgi y que se denominan gránulos de Birbeck. No se conoce su origen y su función no está claramente definida aunque se cree que participan en el transporte intracitoplasmático de antígenos. Desde el punto de vista inmunohistoquímico las células de Langerhans reaccionan con los anticuerpos para la proteína S-100 y para la glicoproteína timocítica CD1. El acúmulo de células de Langerhans es la característica histopatológica esencial para el diagnóstico de HPCL. Las lesiones histológicas progresan desde nódulos celulares con abundantes histiocitos, a nódulos completamente fibróticos; en fases avanzadas de la enfermedad, las lesiones, sobre todo las óseas, puede ser completamente acelulares (3-5).
La incidencia y prevalencia exactas de la HPCL son desconocidas, aunque estudios de biopsias pulmonares en pacientes con enfermedad pulmonar intersticial identifican esta enfermedad en el 5% de los casos, lo que sugiere que es una entidad poco frecuente (3).
No existen asociaciones familiares ni factores genéticos predisponentes aunque esta enfermedad es más frecuente en la raza blanca (6). Tampoco se conocen factores de riesgo geográficos ni ocupacionales que puedan influir en su aparición. La única asociación claramente probada es con el hábito tabáquico. El 90% o más de los pacientes con HPCL son fumadores (4).
La frecuencia relativa hombre/mujer es controvertida. Hace años se creía que esta enfermedad era más frecuente en varones, pero series recientes demuestran una incidencia igual o superior en las mujeres. Esto probablemente sea debido al cambio en los hábitos de tabaquismo en ambos sexos (4).
El 25% de los pacientes están asintomáticos en el momento del diagnóstico. Hasta un 36% de los casos se diagnostican casualmente al hacer una radiología torácica de rutina (7). Los síntomas más frecuentes son la tos no productiva y la disnea. Menos del 5% de los pacientes presentan hemoptisis. Un tercio de los pacientes presentan síntomas constitucionales como pérdida de peso, fiebre, sudor nocturno y anorexia.
El dolor costal puede ser también el síntoma inicial. En algunos casos es debido a una lesión lítica costal, como sucedió en nuestro paciente, pero lo más frecuente es que se deba a un neumotórax (7). El 10-20% de los pacientes debutan con un neumotórax espontáneo, siendo este hecho más frecuente en varones jóvenes.
En el 5-10% de los casos aparecen síntomas debidos a la afectación de otros órganos y entre ellos destacan la diabetes insípida por afectación hipotalámica, el exantema secundario a la infiltración cutánea, las adenopatías por afectación de ganglios linfáticos, o el dolor abdominal por la afectación del hígado o del bazo (3).
La exploración física es generalmente normal, aunque pueden existir alteraciones en la auscultación pulmonar según el estado evolutivo de la enfermedad y el grado de fibrosis que exista.
Los exámenes de laboratorio son inespecíficos.
Las pruebas de función pulmonar pueden mostrar obstrucción, restricción o un patrón mixto. El grado de afectación de la función pulmonar depende de la extensión de la enfermedad y del estadio de la misma. Los volúmenes pulmonares son normales o están incrementados. Este hallazgo es útil para distinguir la HPCL de otras enfermedades intersticiales pulmonares que casi siempre se asocian con volúmenes pulmonares disminuidos (excepto en la linfangioleiomiomatosis, donde los volúmenes están aumentados). Lo más característico es la reducción en la capacidad de difusión del monóxido de carbono que está presente en el 70-90% de los pacientes.
La radiología de tórax es anormal en la mayoría de los pacientes. Los hallazgos que aparecen más precozmente son micronódulos o retículo-nódulos y afectación intersticial. Las lesiones son generalmente bilaterales y simétricas, predominan en los lóbulos superiores y medios y tienden a respetar los ángulos costofrénicos (7,8). Sólo en el 10% de los casos la radiología de tórax es normal.
La TAC de tórax es una técnica útil y sensible para el diagnóstico de HPCL. Lo más frecuente es encontrar en el pulmón, lesiones quísticas menores de 20 mm de diámetro con una pared típicamente fina, de 1 mm de grosor o incluso menos.
Las células de Langerhans pueden ser detectadas en tejido sano y en múltiples patologías pulmonares, por lo tanto su mera presencia no es diagnóstica. El diagnóstico histológico definitivo se basa en la demostración fiable de un incremento de células de Langerhans en las lesiones pulmonares.
La confirmación del diagnóstico puede hacerse mediante lavado broncoalveolar, biopsia transbronquial o biopsia quirúrgica. La presencia de un número aumentado de células de Langerhans en el lavado broncoalveolar, identificadas mediante tinción con anticuerpos frente CD1a, es altamente sugestiva de HPCL. Cuando la proporción es mayor del 5%, el diagnóstico es muy probable. Sin embargo, es frecuente encontrar entre 2-5% de células de Langerhans en el lavado broncoalveolar, incrementos en este rango pueden estar presentes en fumadores importantes o en personas con otras enfermedades intersticiales. La broncoscopia con toma de biopsia transbronquial tiene un bajo rendimiento (10-40%) debido a la distribución parcheada que tienen las lesiones y a la escasa cantidad de tejido que se obtiene. La biopsia quirúrgica (abierta o con toracoscopia) es la prueba con mayor rendimiento diagnóstico, si bien también puede dar falsos negativos debido a que las lesiones pulmonares son focales y se encuentran en diferentes estadios, obteniéndose en estadios avanzados sólo material fibrótico (3).
El abandono del hábito tabáquico es una parte esencial del tratamiento. Con ello se consigue una estabilización e incluso una desaparición, de los síntomas en la mayoría de los pacientes. Hay casos descritos, similares al nuestro, en los que se objetiva una mejoría radiográfica y en la función pulmonar sólo con la abstención tabáquica (9). Los corticoides han sido el pilar del tratamiento médico de esta enfermedad a pesar de que son limitados los estudios que apoyan su eficacia. Parece razonable usarlos en pacientes con enfermedad pulmonar progresiva o con síntomas sistémicos. Se recomienda el tratamiento durante un año con dosis de 0,5-1 mg/kg/día de prednisona o dosis equivalentes de otros glucocorticoides (10). También han sido usados agentes quimioterápicos como vinblastina, metotrexate o ciclofosfamida en pacientes con enfermedad progresiva refractaria a los corticoides o en pacientes con afectación multiorgánica. Se han investigado fármacos que frenan el proceso fibrótico pulmonar como los inmunomoduladores (interferón alfa) y la colchicina (11). El transplante pulmonar es una alternativa para aquellos pacientes con insuficiencia respiratoria avanzada y en casos de hipertensión pulmonar severa, si bien se han descritos recidivas posteriores (10).
La larga y bien documentada serie de Lieberman (12) sitúa a la enfermedad dentro de unos parámetros evolutivos de benignidad; pero es sabido que algunos pacientes pueden evolucionar de forma muy desfavorable hacia una insuficiencia respiratoria severa e incluso hacia la muerte (13). Se han descrito una serie de factores asociados a un peor pronóstico y son las edades extremas de la vida, la afectación multiorgánica, la presencia de síntomas constitucionales, las imágenes radiológicas en panal de abejas, la reducción marcada de la capacidad de difusión y el tratamiento prolongado con corticoides (3). Además el pronóstico puede verse ensombrecido debido a que se ha descrito una mayor incidencia de neoplasias malignas (linfomas, mieloma múltiple, adenocarcinoma de pulmón y otros tumores sólidos) en pacientes con HPCL, aunque esto puede ser una asociación casual (3,7).
En conclusión, hemos considerado interesante la presentación de este caso de HPCL ya que su descripción ilustra los hallazgos clínicos, radiológicos e histopatológicos que caracterizan a esta entidad. Destacamos también el hecho de que la HPCL es una enfermedad poco frecuente y de difícil diagnóstico porque puede ser asintomática, porque hay pacientes que presentan una remisión espontánea y porque la biopsia pulmonar en fases avanzadas puede no ser diagnóstica.
Bibliografía
1. Lichtenstein L. Histiocytosis X: integration of eosinophilic granuloma of bone, Letterer- Siwe disease and Schüller Christian disease, as related manifestations of a single nosologic entity. AMA Arch Pathol 1953; 56: 84-102. [ Links ]
2. Morell F, Reyes L, Majó J, Orriols R. Histiocitosis de células de Langerhans. Estudio clínico y evolución a largo plazo de 21 pacientes. Med Clin (Barc) 2000; 115: 60-64. [ Links ]
3. Videragay F, Martínez M, Maldonado P, Zulaica H, Vázquez E. Histiocitosis de células de Langerhans. Ann Med Asoc Med Hosp ABD 2002; 47 (1): 38-43. [ Links ]
4. Vassallo R, Ryu JH, Colby TV, Hartman T, Limper AH. Pulmonary Langerhans'-cell histiocytosis. N Engl J Med 2000; 342 (26): 1969-78. [ Links ]
5. Pere B, Mallofré C, Pomés J. Varón de 20 años de edad con dolor lumbosacro y lesiones osteolíticas. Med Clin (Barc) 2001; 117: 309-15. [ Links ]
6. Friedman PJ, Liebow AA, Sokoloff J. Eosinophilic granuloma of lung: clinical aspects of primary histiocytosis in the adult. Medicine (Baltimore) 1981; 60: 385-96. [ Links ]
7. Tazi A, Soler P, Hance AJ. Adult pulmonary Langerhans'cell Histiocitosis.Thorax 2000; 55: 405-16. [ Links ]
8. Bonelli FS, Hartman TE, Swenson SJ, Sherrick A. Accuracy of high resolution CT in diagnosing lung diseases. Am J Roentgenol 1998; 170: 1507-12. [ Links ]
9. Mogulkoc N, Veral A, Bishop PW, Bayindir U, Pickering CA, Egan JJ. Pulmonary Langerhans' cell histiocytosis: radiologic resolution following smoking cessation. Chest 1999; 115: 1452-55. [ Links ]
10. Abellán Martínez MC, Méndez Martínez P, Sánchez Gascón F, Hernández Martínez J, Sánchez Montón T, Romero Mas E. Histiocitosis X pulmonar. Presentación de un caso y revisión de la literatura. An Med Interna (Madrid) 2000; 19: 28-30. [ Links ]
11. Giona F, Caruso R, Testi AM, et al. Langerhans' cell histiocytosis in adults: a clinical and therapeutic analysis of 11 patients from a single institution. Cancer 1997; 80: 1786-91. [ Links ]
12. Lieberman PH, Steinman RM, Smith J, Garin-Chesa P, Urmacher C, Sperber M. Langerhans'cell (eosinophilic) granulomatosis. Am J Surg Pathol 1996; 20: 519-552 . [ Links ]
13. Orriols R, Bravo C, Bernardó L, Tallada N, Mesa J, Morell E. Histiocitosis X con afección pulmonar. Estudio de catorce casos. Med Clin (Barc) 1988; 91: 365-370. [ Links ]
![]() Dirección para correspondencia
Dirección para correspondencia
Ana María García Bernárdez
Servicio de Medicina Interna I
Hospital Universitario Central de Asturias
C/ Celestino Villamil, s/n.
33006 Oviedo.
Trabajo aceptado: 12 de septiembre de 2005.

