










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.24 no.5 may. 2007
Enfermedad de Lhermitte-Duclos asociada a enfermedad de Cowden: a propósito de un caso
Lhermitte-Duclos´s disease associated to Cowden´disease: a case report
I. Lavín Castejón, J. Mut Oltra1, C. Trillo Balizón C, A. Maldonado Barrionuevo2
Servicio Aparato Digestivo. Hospital Clínico Virgen de la Victoria.
1Unidad de Dermatología.
2Servicio de Medicina Interna. Hospital de la Axarquía. Vélez. Málaga
Dirección para correspondencia
RESUMEN
La enfermedad de Cowden es una rara genodermatosis que se caracteriza por la aparición de multiples hamartomas tanto a nivel cutaneomucoso como visceral. Es una lesión cerebelosa que consiste en el engrosamiento displásico de las circunvoluciones cerebelosas. Se incluye dentro de las facomatosis y suele presentarse junto a la enfermedad de Cowden, la esclerosis tuberosa o síndromes de solapamiento.
Presentamos un paciente de 56 años diagnosticado en la consulta de Dermatología de enfermedad de Cowden hace 10 años. En el estudio de extensión se le diagnostica de poliposis intestinal hamartomatosa, acantosis glucogénica esofágica y se encuentran dos nódulos sólidos tiroideos. A los 10 años del diagnóstico se realiza resonancia magnética nuclear cerebral por aparición de cefalea encontrando una masa mal definida en hemisferio cerebeloso derecho compatible con gangliocitoma displásico cerebeloso.
Palabras clave: Enfermedad de Lhermitte-Duclos. Gangliocitoma displásico Enfermedad de Cowden. Hamartomatosis múltiple. Poliposis gastrointestinal. RMN.
ABSTRACT
Cowden's disease is a rare genodermatosis that is characterized for multiple cutaneus and visceral hamartoma . Lhermitte-Duclos´s disease is a cerebelous lesion that consists in the displasic enlargement of the cerebelous circumvolution. It's incluyed in phacomatosis and ussually presents associated to Cowden's disease, tuberous sclerosis and overlap syndromes.
A 56 years old man, diagnosed in Dermatology with Cowden's disease ten years ago. In the extension study, he had hamartoma intestinal polip, esophagic glucogenic acanthosis and two solid thyroid nodules. The craneal TC didn't show significant alteration. Ten years after diagnosis a cerebral magnetis resonance was performanced for intense cephalea, and it was found a bad-defined mass in right cerebelous hemisphere without contrast captation, compatible with cerebelous glangliocytoma.
Key words: Lhermitte-Duclos´s disease. Displasic gangliocytoma. Cowden's disease. Multiple hamartomatosis. Gastrointestinal poliposis. MRN.
Introducción
La enfermedad de Lhermitte-Duclos o gangliocitoma displásico cerebeloso es un tumor unilateral, lentamente progresivo, que consiste en un engrosamiento displásico de las circunvoluciones cerebelosas. Se incluye dentro del espectro de las facomatosis y suele asociarse a la enfermedad de Cowden. Aproximadamente hay publicados 220 casos de Lhermitte-Duclos en total (1). En un estudio multicéntrico realizado en 8 hospitales franceses con departamento de dermatología se realizaron resonancias magnéticas cerebrales a 20 pacientes con enfermedad de Cowden encontrándose anomalías en el 35% de los casos (tres L'hermitte-Duclos, un meningioma y seis malformaciones vasculares) (2). También se encuentra en la literatura asociada a otras enfermedades como la esclerosis tuberosa, neurofibromatosis tipo I o a síndromes de "solapamiento" (3,4).
La enfermedad de Cowden es una enfermedad hereditaria extremadamente rara (menos de 500 casos publicados) que se caracteriza por la presencia de lesiones mucocutáneas características y diagnósticas, asociadas con lesiones hamartomatosas viscerales y la formación de neoplasias malignas, fundamentalmente mamarias y tiroideas, en la edad adulta (5). Se hereda como un rasgo autosómico dominante, de penetrancia variable, habiéndose identificado el gen relacionado en el brazo largo del cromosoma 10 (10q23.31, 10q22.3), llamado gen PTEN (phosphatase and tensin homologue) que actúa normalmente como gen supresor tumoral (6-8).
La enfermedad de Cowden afecta por igual a ambos sexos y aparece en la segunda o tercera década de la vida (9). Se reconoce clínicamente por la presencia de lesiones mucocutáneas típicas, que aparecen en el 80% de los pacientes, siendo la de mayor interés para el diagnóstico los llamados triquilemomas o tricolemomas (lesiones múltiples que corresponde a tumores benignos del folículo piloso), que se manifiestan como pápulas faciales, de color carne y se agrupan especialmente alrededor de boca nariz o pabellones auriculares, cuando se agrupan dan lugar a una imagen típica en empedrado.
Otro de los rasgos clínicos es la presencia de hamartomas o fibroadenomas múltiples, siendo típicas la aparición de hamartomatosis visceral: tiroidea, mamaria y a cualquier nivel del aparato reproductor femenino, poliposis gastrointestinal y del sistema nervioso central, hemangiomas, neuromas, y en algunos casos estrías angioides en la retina (9,10).
Existe un elevado riesgo de malignización de los fibroadenomas mamarios en las mujeres y de los tiroideos en los hombres, incluso a edad temprana, también puede aparecer carcinoma a nivel del tracto gastrointestinal (10,11).
Caso aportado
Varón de 56 años diagnosticado hace 10 años de Enfermedad de Cowden por presentar múltiples formaciones papilomatosas en dorso de la lengua de tamaño variable, macroglosia saburral y pápulas de color piel en mucosa lingual a modo de empedrado. El resultado anatomopatoógico fue de pólipos fibroepiteliales (12) (Fig. 1).
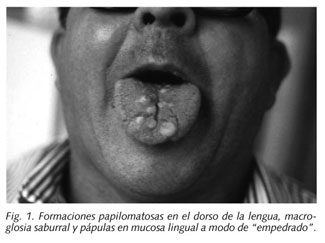
Tras el diagnóstico dermatológico se inició el estudio de extensión: a) tránsito EGD y enema opaco que evidencian numerosos pólipos desde el antro hasta la ampolla rectal; b) endoscopias digestivas alta y baja: parcheado difuso desde esófago cervical a cardias que histológicamente coincide con una acantosis glucogénica; pólipos sésiles y alguno pediculado desde estómago hasta ampolla rectal que corresponden histológicamente a pólipos hamartomatosos; c) ecografía tiroidea: 2 nódulos sólidos, uno en cada lóbulo. Tras punción (PAAF) se diagnostica de bocio coloidal; y d) se realizó tomografía computerizada craneal que no demostró alteraciones significativas.
En la actualidad el paciente consulta por cefalea. Dada su enfermedad de base se le realiza resonancia magnética nuclear demostrándose una lesión en hemisferio cerebeloso derecho descrita como un aumento de volumen debido a la presencia de una masa mal delimitada que muestra discreta hipodensidad de señal T1 y discreta hiperdensidad de señal T2 y FLAIR que afecta a la cortical cerebelosa en región posterolateral y que condiciona un leve desplazamiento hacia la izquierda del IV ventrículo sin evidencia de hidrocefalia a nivel supratentorial. Tras la administración de contraste paramagnético no se visualiza captación significativa por lo que parece tratarse de un gangliocitoma displásico cerebeloso con engrosamiento de las capas del cerebelo lo que produce un aparente laminado de la cortical (Fig. 2).
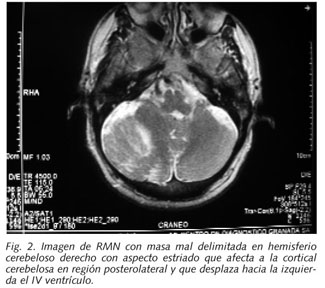
Discusión
El gangliocitoma displásico cerebeloso se presenta habitualmente entre los 20-40 años. Suele manifestarse con cefalea por hipertensión intracraneal e hidrocefalia y ocasionalmente vértigo. Puede ocurrir además hipotensión ortostática y manifestaciones psiquiátricas. Los hallazgos de la tomografía axial son inespecíficos, como una lesión hipodensa de límites escasamente definidos y que puede protuir hacia el IV ventrículo. Sin embargo la resonancia magnética constituye el método de diagnóstico. Se manifiesta por una masa cerebelosa con aspecto estriado en forma de bandas hiperintensas en las secuencia T2 e hipodensas en las T1. Hay publicaciones en las que el diagnóstico de L'hermitte-Duclos se ha realizado mediante tomografía por emisión de positrones con 11C-metionina (13).
El caso que nos ocupa viene a confirmar la asociación entre gangliocitoma displásico cerebeloso y la enfermedad de Cowden, considerándose la enfermedad de L'hermitte-Duclos un criterio mayor para el diagnóstico de la enfermedad de Cowden (14).
El diagnóstico de la enfermedad de Cowden es fundamentalmente clínico, habiéndose establecido unos criterios diagnósticos por el Consorcio Internacional del Síndrome de Cowden, que tras la revisión efectuada en el año 2000, quedan definidos como se puede apreciar en la tabla I (1,2,9,10).
El diagnóstico precoz del síndrome es de gran importancia, pues un diagnóstico de síndrome de Cowden, siempre conlleva un despistaje de los tumores malignos, de aparición juvenil con los que frecuentemente se asocia. El carcinoma de mama, generalmente bilateral, afecta al 30% de las mujeres, por lo general con una edad media al diagnóstico de 40 años; el carcinoma de tiroides afecta al 7% de los pacientes, preferentemente hombres; menos frecuentes son los tumores malignos del tracto gastrointestinal, sobre todo colon y pueden aparecer de forma excepcional tumores malignos en pulmón, útero, vejiga y sistema hematopoyético (15,16).
Una vez diagnosticado el paciente según los criterios dermatológicos es necesario hacer seguimiento de las manifestaciones viscerales asociadas, en nuestro paciente destacan las manifestaciones digestivas, tiroideas y neurológicas, controlando posibles malignizaciones. Además ante la aparición de clínica neurológica se debe realizar resonancia magnética para buscar posible hamartoma cerebeloso. En la tomografía computerizada realizada 10 años antes no se encontró ninguna anomalía, dado que no existía hidrocefalia. El diagnóstico debe realizarse mediante resonancia magnética nuclear. En el caso que nos ocupa el gangliocitoma displásico cerebeloso no se ha tratado por ahora. En el caso de aparecer hidrocefalia hipertensiva se realizaría una derivación ventrículo-peritoneal (3).
No existe tratamiento curativo para la enfermedad, aunque el control evolutivo de las lesiones asociadas permitirá la cirugía antes de que ocurran malignizaciones.
Bibliografía
1. Robinson S, Cohen AR. Cowden disease and Lhermitte-Duclos disease: An update. Case report and review of the literature. Neurosurg Focus 2006; 20 (1): E6. [ Links ]
2. Lok C, Viseux V, Avril MF, Richard MA, Gondry-Jouet C, Deramond H, et al. Cancerology Group of the French Society of Dermatology. Brain magnetic resonance imaging in patients with Cowden syndrome. Medicine (Baltimore) 2005; 84 (2): 129-36. [ Links ]
3. Ortega R, Escamilla F, Pastor J, Romero F, Mínguez A. Enfermedad de Lhermitte-Duclos asociada a esclerosis tuberosa. Presentación de un caso y revisión de la literatura. Rev Neurol 2000; 30, 9: 833. [ Links ]
4. Yesildag A, Baykal B, Ayata A, Kerman G, Koroglu M, Olgar S, et al. Lhermitte-Duclos disease associated with neurofibromatosis type-1 and non-ossifying fibroma. Acta Radiol 2005; 46 (1): 97-100. [ Links ]
5. Fistarol SK, Anliker MD, Itin PH. Cowden disease or multiple hamartoma syndrome cutaneous clue to internal malignancy. Eur J Dermatol. 2002; Sep-Oct; 12 (5): 411-21. [ Links ]
6. Reifenberger J, Rauch L, Beckmann MW, Megahed M, Ruzicka T, Reifenberger G. Cowden's disease: Clinical and molecular genetic findings in a patient with a novel PTEN mutation. Br J Dermatol 2003 May; 148 (5): 1040-6. [ Links ]
7. Abel TW, Baker SJ, Fraser MM, Tihan T, Nelson JS, Yachnis AT, et al. L'hermitte-Duclos disease: A report of 31 cases with inmunohistochemical analysis of the PTEN/AKT/mTOR pathway. J Neuropathol Exp Neurol 2005; 64 (4): 341-9 [ Links ]
8. Blanco V, Keochgerian V. Cowden's syndrome. Case report, with reference to an affected family. Med Oral Patol Oral Cir Bucal 2006; 11 (1): 12-6 [ Links ]
9. Yañez S, Martín-Santiago A, Mestre F, González A, Pinazo I. Enfermedad de Cowden. Actas Dermo-Sif 1992; 83, 11: 587-90. [ Links ]
10. Starink M. Cowden's disease: Analysis of fourteen new cases. J Am Acad Dermatol 1984; 11: 1127-41. [ Links ]
11. Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous popyposis syndromes: A clinical and moleculare review. Am J Gastroenterol 2005; 100 (2): 476-90 [ Links ]
12. Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: Case report with human papillomavirus studies. J Am Acad Dermatol 2004; 51 (3): 411-5. [ Links ]
13. Calenbergh FV, Vantomme N, Flamen P, Daerel Ph, Sciot R, Legius E, et al. L'hermitte-Duclos disease: 11 C-methionine positron emission tomography data in 4 patients. Surgical Neurology 2006 ; 65: 293-7. [ Links ]
14. Boonpipattanapong T, Phuenpathom N, Mitarnun W. Cowden's syndrome with L'hermitte-Duclos disease. Br J Neurosurg 2005; 19 (4): 361-5. [ Links ]
15. Fistarol SK, Anliker MD, itin PH. Cowden disease or multiple hamartoma syndrome cutaneous clue to internal malignancy. Eur J Dermatol 2002; 12 (5): 411-21. [ Links ]
16. Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: A clinical and molecular review. Am J Gastroenterol 2005; 100 (2): 476-90. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Isabel Lavín Castejón.
C/ Centaurea, 7, escalera C, 5B.
29018 Málaga.
e-mail: med008977@saludalia.com
Trabajo aceptado: 28 de diciembre de 2006

