










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.25 no.2 feb. 2008
Hemoglobinopatías diagnosticadas en el área sanitaria del Hospital Universitario Nuestra Señora de Candelaria de Santa Cruz de Tenerife durante un año
Hemoglobinopathies diagnosed at the University Hospital Ntra. Sra. de Candelaria and its area of reference in Santa Cruz de Tenerife during one year
S. de las Heras Flórez, L. M. Pérez Hernández
Servicio de Análisis Clínicos. Hospital Universitario Nuestra Señora de Candelaria. Santa Cruz de Tenerife
Dirección para correspondencia
RESUMEN
Objetivo: Describir número y tipo de hemoglobinopatías diagnosticadas en nuestro área de referencia durante un año así como el origen de estos pacientes, para tener un primer contacto con esta patología hasta ahora no caracterizada en nuestra provincia.
Material y método: Se revisaron las hemoglobinopatías diagnosticadas durante un año, bien por sospecha clínica, al realizar el cromatograma para la cuantificación de hemoglobina (Hb) A1c o por sospecha de β-talasemia en el hemograma. El método de despitaje ha sido la cromatografía líquida de alta resolución (HPLC), que identifica y cuantifica HbA2, HbS y HbC. El resto de variantes fueron diagnosticadas en el laboratorio de referencia.
Resultados: Se diagnosticaron 198 hemoglobinopatías, 105 a partir de la realización de HbA1c. Los diagnósticos más frecuentes fueron β-talasemia minor y HbS en estado heterocigoto. El 61,1% de los pacientes son autóctonos de Canarias, mayoritariamente de Tenerife.
Conclusiones: El alto número de pacientes autóctonos de Canarias portadores de hemoglobinopatías encontrado, nos indica que éste debe ser el punto de partida en el diseño de estudios prospectivos sobre grupos poblacionales concretos para conocer la prevalencia, y plantear el desarrollo de programas de control y prevención en la provincia de Santa Cruz de Tenerife.
Palabras clave: Hemoglobinopatías. Santa Cruz de Tenerife.
ABSTRACT
Objective: To describe number and type of hemoglobinopathies diagnosed in our area of reference during one year as well as the origin of these patients, to have a first contact with this pathology not characterized in our province until now.Material and method: All hemoglobinopathies diagnosed in one year were reviewed, either for suspected patients, after analyzing chromatogram to quantify hemoglobin (Hb) A1c or hemogram suspicious of β-thalassemia. The screening method was high pressure liquid chromatography (HPLC), to identify and quantify HbA2, HbS and HbC. The other variants were diagnosed at reference laboratory.
Results: 198 hemoglobinopathies were diagnosed, 105 after measure HbA1c. The more frequent diagnoses were β-thalassemia trait and heterozygote HbS. 61.1% of patients are from Canary Islands, mainly from Tenerife.
Conclusions: The high number of carriers found from Canary Islands, should be the start to design prospective studies on population groups, in order to know the hemoglobinopathies prevalence and to perform control and prevention official programs in the province of Santa Cruz de Tenerife.
Key words: Hemoglobinopathies. Santa Cruz de Tenerife.
Introducción
Las hemoglobinopatías son las alteraciones monogénicas más frecuentes en el mundo y tienen un patrón de herencia autosómico recesivo. Se dividen en: hemoglobinopatías estructurales, producidas por la síntesis de una cadena de globina estructuralmente anormal, y síndromes talasémicos, que se manifiestan por la disminución total o parcial de la síntesis de una globina de estructura normal (1).
INCIDENCIA Y DISTRIBUCIÓN GEOGRÁFICA
La frecuencia de hemoglobinopatías en la población mundial es muy elevada y su distribución geográfica muy variable. Se calcula que cada año nacen en todo el mundo más de 300.000 niños con formas graves de estas enfermedades, la mayoría de ellos en países de ingresos bajos y medios. Aproximadamente un 5% de la población mundial es portadora de un gen de la drepanocitosis o de la talasemia. El porcentaje de portadores puede alcanzar el 25% en algunas regiones (2). Aproximadamente el 60-70% de todos los nacimientos de niños con alguna alteración grave de la hemoglobina (Hb) se producen en África, siendo la región subsahariana la más afectada. Aunque globalmente existen más portadores de talasemia, la elevada frecuencia de presentación del gen falciforme (HbS) en ciertas áreas conlleva la aparición de un alto número de nacimientos de individuos con drepanocitosis. Debido al aumento de los fenómenos migratorios, estas enfermedades están apareciendo con mayor frecuencia en muchas zonas no endémicas (3).
Las hemoglobinopatías estructurales más ampliamente extendidas por toda la geografía mundial son la HbS, HbC, HbE y HbD Punjab. La HbS es la más frecuente, afectando sobre todo a África tropical y subtropical. Le siguen en frecuencia las HbC, HbD y HbE, que se presentan en poblaciones del oeste de África, India y suroeste de Asia.
Las talasemias tienen una frecuencia variable según las zonas geográficas, presentando una especial predilección por las poblaciones del área mediterránea, con predominio de la β-talasemia y por las de Oriente Medio, Sudeste Asiático y China, con predominio de la α-talasemia (3).
Situación en España: no existe información contrastada sobre la verdadera incidencia o prevalencia de las diferentes formas de hemoglobinopatías, no existiendo un registro oficial de estos pacientes ni en edad infantil ni adulta.
En cuanto a las hemoglobinopatías estructurales, la mayoría de estudios en España corresponden a investigaciones descriptivas en regiones geográficas concretas. En el estudio de Dulín Iñiguez et al (4) realizado en 2003 en neonatos de la Comunidad de Madrid, la incidencia global de variantes de hemoglobinas fue de 1/299 recién nacidos analizados, con una incidencia de anemia falciforme de 1/5.851 casos analizados. En el estudio de Cabot et al (5) realizado en 1998 en Mataró en 82 neonatos de raza negra, la prevalencia de portadores de HbS fue de 10,98‰ y en estado homocigoto de 1,22‰. En Gran Canaria, Malcorra et al (6) en 1994, encontraron una tasa de prevalencia de hemoglobinopatías estructurales del 2,7‰. En el estudio de Calvo-Villas (7), realizado en Lanzarote en 2005, se analizaron 2.436 gestantes, encontrando que el 9,44‰ presentaban hemoglobinopatías estructurales en estado heterocigoto.
Respecto a las talasemias, en un estudio efectuado en 1986 por el Grupo de Estudio de Hemoglobinopatías y Talasemias (GEHBTA) sobre 25.000 individuos de diferentes regiones de España, se detectó una amplia heterogeneidad en la tasa de prevalencia de esta enfermedad, que oscila desde la ausencia de la misma en población vasca hasta el 5% detectado en la isla de Menorca (8). La prevalencia global promedio en España se ha estimado en el 0,4%, que corresponde a un caso de β-talasemia por cada 250 habitantes (9). En otros estudios limitados a áreas concretas del país, como el de Malcorra et al realizado en 1993 (10) en 250 individuos adultos sanos de la isla de Gran Canaria, referido a la población hindú de origen Shindi, se encontró la mayor tasa de prevalencia de β-talasemia registrada en España con un 10,5%. El estudio publicado por Oliva et al en 1998, realizado en población escolar de Menorca, refleja una prevalencia de β-talasemia del 2,67% (11).
Desde finales de la década de los 80 se ha producido una importante inmigración a España y otros países europeos, de habitantes de las regiones más deprimidas y con menos expectativas del planeta (África subsahariana, Zagreb, Latinoamérica, sureste asiático y este de Europa), concentrándose especialmente en los grandes núcleos urbanos. Todos estos cambios étnicos pueden producir el desarrollo de una gran heterogeneidad de patologías y lesiones moleculares de la hemoglobina, con la aparición de variantes estructurales en poblaciones carentes de ellas o cuya frecuencia era muy baja antes de la emigración transcontinental.
Debido al desconocimiento de la prevalencia de hemoglobinopatías en nuestra población de referencia y al progresivo aumento de fenómenos migratorios desde países con altas tasas de prevalencia de estas enfermedades, se planteó diseñar un estudio para conocer el número y tipo de hemoglobinopatías diagnosticadas durante el periodo de 1 año en nuestro área de referencia así como conocer el lugar de nacimiento de estos pacientes, y de esta manera, tener una primera toma de contacto con esta patología hasta ahora no caracterizada en nuestra provincia.
Material y método
Las muestras analizadas proceden de nuestro área de referencia, que comprende una población de 481.061 habitantes, pertenecientes al núcleo urbano de Santa Cruz de Tenerife, la zona sur de la isla de Tenerife, así como a las islas de La Gomera y El Hierro (12). El periodo de estudio comprende desde Septiembre 2005 a Septiembre de 2006. Los datos fueron obtenidos del sistema informático OpenLab.
Se estudiaron todas las muestras remitidas al laboratorio solicitando estudio de posible hemoglobinopatía por sospecha clínica, analítica o en el contexto de un estudio familiar. También se seleccionaron las muestras en las que se realizó hemoglobina glicada (HbA1c) y que al revisar el cromatograma presentasen cualquier alteración que pudiera hacer sospechar una hemoglobinopatía. Asimismo se seleccionaron los hemogramas con resultados sospechosos de β-talasemia: volumen corpuscular medio de hematíes disminuido y poliglobulia, acompañado de alteraciones en el frotis como punteado basófilo de hematíes o dianocitos.
En todas estas muestras, el despistaje de hemoglobinopatías se ha realizado mediante cromatografía líquida de alta resolución (HPLC), en un analizador Menarini 8160. Este equipo, realiza una cromatografía de 240 segundos, que permite cuantificar HbA2 y HbFetal. También identifica y cuantifica HbS y HbC en una misma ventana. Para su posterior identificación se ha realizado el test de solubilidad: un resultado positivo indica HbS y uno negativo HbC.
En los casos en los que ha sido necesario, ante la sospecha de otras variantes de Hb diferentes a la S o C, se han remitido las muestras a un laboratorio de referencia para completar el estudio (mediante cromatografía de mayor duración y/o electroforesis) y así llegar al diagnóstico. En la figura 1 se detalla el procedimiento empleado en este laboratorio en el diagnóstico de hemoglobinopatías.

Debido a que los portadores de α-talasemia no se detectan en la cromatografía, no están incluidos en el presente estudio.
Se recogieron en una base de datos edad, sexo y origen de todos los pacientes, registrando tanto su lugar de nacimiento como el origen de sus progenitores. En los pacientes de origen canario se recogió la isla de procedencia, del resto de España su comunidad autónoma y de otras nacionalidades el país de origen. Se analizaron los datos con el paquete estadístico SPSS versión 14.0.
Resultados
Durante el período de estudio se diagnosticaron 198 hemoglobinopatías: 105 a partir del hallazgo de un cromatograma anormal al realizar la HbA1c, 53 en el contexto de un estudio de posible hemoglobinopatía y 40 por sospecha de talasemia en el hemograma. La edad media al diagnóstico fue de 45 años (percentil 25-75: 30-64; rango: 1-92). El 57% fueron mujeres.
Las hemoglobinopatías diagnosticadas fueron: 71 β-talasemias minor, 2 β-talasemias intermedias, 70 HbS en estado heterocigoto (HbA/S), 2 HbS en estado homocigoto (HbS/S), 1 doble heterocigoto β-talasemia/HbS, 30 HbD en estado heterocigoto (HbA/D), 18 HbC en estado heterocigoto (HbA/C), 2 HbG en estado heterocigoto (HbA/G), 1 HbE en estado heterocigoto (HbA/E) y 1 HbJ Birmingham en estado heterocigoto (HbA/J).
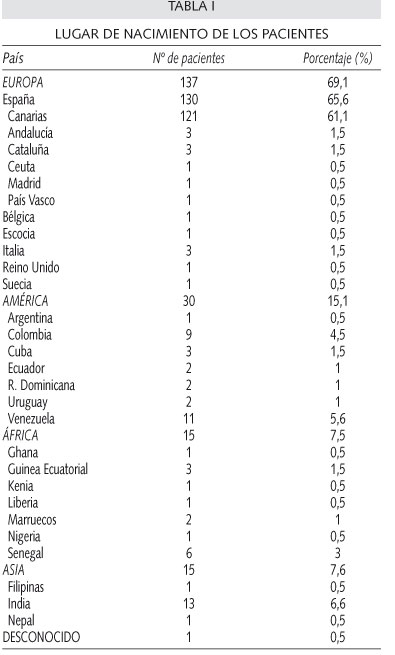
Globalmente, 121 pacientes son nacidos en Canarias (61,1%) y 9 en otras comunidades (4,5%), 7 en el resto de Europa (3,5%), 15 en África (7,5%), 30 en América (15%), 15 en Asia (7,5%) y en 1 caso no se pudo conocer el origen (0,5%). Los países de nacimiento más frecuentes después de España (65,6%) fueron: India (6,6%), Venezuela (5,5%), Colombia (4,5%) y Senegal (3%). De los 102 canarios cuya isla de procedencia pudo ser recogida, el 83,3% son de Tenerife.
Analizando el origen de los pacientes atendiendo a la hemoglobinopatía diagnosticada, la mayoría de los pacientes diagnosticados de β-talasemia minor (60,6%), HbA/S (60%) y HbA/D (86,7%) son nacidos en Canarias, destacando también los 9 nacidos en la India con talasemia minor y los 10 de África subsahariana con HbS. Los diagnosticados de HbA/C son originarios del continente americano y de España en igual proporción, y tanto los dos diagnósticos de HbA/G como el de HbA/E son originarios de la India. El paciente doble heterocigoto HbS/β-talasemia nació en Canarias y con progenitores canarios y uno de los homocigotos para la HbS nació en Canarias con progenitores de Canarias y Cuba. En las tablas I y II se recogen los lugares de nacimiento por islas si es en Canarias, por comunidades autónomas si es el resto de España y por países para los demás orígenes.
Si tenemos en cuenta cómo fueron diagnosticadas las hemoglobinopatías, tenemos 3 grupos: el grupo 1 diagnosticado a partir de la realización de la HbA1c, el grupo 2 por sospecha del clínico y el grupo 3 a partir del hemograma. La edad media del grupo 1 es de 53,7 años, la del grupo 2 de 29,1 años y la del grupo 3 de 45,4 años. Durante el periodo estudiado recibimos 137 peticiones para realización de estudio de hemoglobinopatías por parte de los clínicos, de las cuales se confirmaron 53 (38,6%) y realizamos HbA1c en 45.130 pacientes, diagnosticándose 105 hemoglobinopatías.
En cuanto a las hemoglobinopatías diagnosticadas en cada grupo y el origen de estos pacientes encontramos: en el grupo 1 el diagnóstico más frecuente fue HbA/S (54 pacientes: 51,4%) seguido de HbA/D (24 pacientes: 22,9%) y en el grupo 2 fue la talasemia minor (25 pacientes: 47,2%). En los 3 grupos la mayoría de los pacientes eran nacidos en Canarias (58,1% en el grupo 1, 71,7% en el grupo 2 y 55% en el grupo 3), destacar también 25 pacientes nacidos en América en el grupo 1. En las tablas III, IV y V se detallan los diagnósticos y origen de los pacientes de estos 3 grupos. Los 2 pacientes con HbS/S y el doble heterocigoto β-talasemia/HbS fueron diagnosticados por sospecha del clínico en los primeros meses de vida.



Además de recoger el lugar de nacimiento interrogamos sobre el origen de los padres de los pacientes; en su mayor parte (94%) procedían de las mismas áreas geográficas. Sin embargo; cabe destacar el hallazgo de padres españoles de pacientes nacidos en otros países, como fue el caso del paciente diagnosticado de HbA/S nacido en Filipinas (con padres originarios de Murcia), de los diagnosticados de HbA/D nacidos en Marruecos (con padres de Granada) y Argentina (con padres de Galicia), y del diagnosticado de HbA/C nacido en Venezuela (con padres de Tenerife).
El 16,6% de los pacientes diagnosticados en este estudio son familiares de primer grado, pertenecientes a 15 núcleos familiares diferentes.
Discusión
Las hemoglobinopatías estructurales y las talasemias constituyen un problema de salud pública en algunas zonas geográficas y debido a los movimientos migratorios de las últimas décadas, se les está prestando cada vez más atención en los países occidentales, donde hasta entonces tenían una prevalencia muy baja. Con vistas a disminuir la morbilidad y mortalidad en los primeros años de vida de estos pacientes, en España ya se está realizando cribado neonatal de estas patologías de forma universal en las comunidades de Madrid y Extremadura y en población de riesgo en Cataluña (13-15), y en Galicia se ha emitido un informe favorable sobre la relación coste efectividad de un cribado selectivo en poblaciones con mayor riesgo de presentar hemoglobinopatías (16).
En la actualidad no existe ningún programa de prevención y control de las hemoglobinopatías en nuestro área sanitaria. En nuestro laboratorio, cuando se diagnostica una hemoglobinopatía (Fig. 1), los informes van acompañados del consejo al clínico de ampliar el estudio a los familiares de primer grado. Con esta sistemática, se ha observado que con relativa frecuencia se diagnostican hemoglobinopatías y por tanto se plantea la necesidad de conocer el número y tipo de hemoglobinopatías diagnosticadas así como el origen de estos pacientes y la posible influencia de los movimientos migratorios en su aparición.
Al analizar los resultados de este estudio, se puede observar cómo las hemoglobinopatías más diagnosticadas han sido la HbS en estado heterocigoto y la β-talasemia minor, datos coincidentes con los encontrados en otras áreas del país (4,7).
Como en otras áreas de España, en la Comunidad Canaria y más en concreto en la isla de Tenerife, ha habido un importante aumento de la población en los últimos años, debido a la llegada de habitantes de países en vías de desarrollo sobre todo de América y África y también de otras comunidades españolas. Canarias es actualmente una de las 7 comunidades españolas con más de un 10% de población extranjera (17). En cuanto a la isla de Tenerife, hay un 16,72% de población extranjera, de los cuáles el 7,9% provienen de América (sobre todo Venezuela, Argentina, Cuba y Colombia), y un 1% de África (sobre todo de Marruecos, Senegal y Mauritania), además el 8,3% de la población censada ha nacido en otra comunidad del país. También es de destacar la importante colonia hindú afincada en Tenerife. En lo que respecta a las islas de La Gomera y El Hierro, la proporción de población inmigrante es menor que en la isla de Tenerife con cifras en torno al 1,4% (18).
Estos movimientos migratorios pueden explicar las hemoglobinopatías encontradas en población extranjera, sin embargo la mayoría de hemoglobinopatías diagnosticadas en este estudio ha sido en pacientes originarios de la comunidad Canaria y más en concreto de la isla de Tenerife, lo que difiere de lo publicado recientemente en otras áreas de España en lo que a estructurales se refiere. Tanto en la evaluación del cribado neonatal de la Comunidad de Madrid en 2006 (4), como en el estudio en gestantes Lanzarote en 2006 (7) y en el estudio piloto de cribado neonatal de Cataluña de 2005 (15) se ha encontrado que la mayoría de hemoglobinopatías diagnosticadas ha sido en población inmigrante, originaria de países en vías de desarrollo.
Creemos que el hecho de que la mayoría de hemoglobinopatías se encuentren en población no inmigrante, pueda estar motivado por el origen de los habitantes del archipiélago y su relación con África, como ya apuntaba Malcorra (10) en su estudio realizado en Gran Canaria, en el que encontró tasas de prevalencia de hemoglobinopatías estructurales mayores a las existentes en el resto del país en ese momento, previo a los movimientos migratorios, que atribuyó a la mezcla de inmigrantes europeos originarios de España, Portugal, Italia y Francia con la población nativa de las islas de origen desconocido (guanches) y con un gran número de individuos africanos de raza negra que llegaron como esclavos en los siglos XVI y XVII al archipiélago.
Por otra parte, esta elevada proporción de pacientes originarios de las islas puede deberse al tipo de pacientes seleccionados, ya que más de la mitad son controles de diabetes, y los inmigrantes suelen ser una población joven y por tanto con poca incidencia de esta enfermedad. Pese a esto, en nuestra opinión, las hemoglobinopatías diagnosticadas en población autóctona representan un número considerable y para nada desdeñable.
De los 3 grupos estudiados, el grupo 1, de pacientes en control por diabetes, podría ser considerado como representativo de nuestra población ya que no presentan una sospecha previa de hemoglobinopatía, en contraposición a lo que ocurre en los otros 2 grupos. Teniendo en cuenta a los 45.130 pacientes a los que se realizó HbA1c en el periodo de estudio, existiría una prevalencia de hemoglobinopatías del 2,3‰, siendo prácticamente todas ellas estructurales, con una prevalencia de portadores de HbS del 1,2‰, cifras inferiores a las encontradas en estudios recientes en otras áreas del país (4,7). Sin embargo, el considerar este grupo de pacientes como representativo de nuestra población tiene una serie de limitaciones. En primer lugar, como ya se apuntó previamente, la HbA1c se realiza para el control y diagnóstico de diabetes, y la incidencia de esta enfermedad aumenta con la edad. La edad media de este grupo es de 53,7 años, por lo tanto, no se están considerando los grupos poblacionales más jóvenes en los cuales se sitúan la mayoría de los inmigrantes. En segundo lugar, solamente se estudian las hemoglobinopatías no conocidas previamente, y este hecho puede explicar que la mayoría sean estructurales heterocigotas y por tanto inadvertidas para el clínico. La baja proporción de β-talasemias diagnosticadas en este grupo, puede ser debida a que probablemente estos pacientes ya fueron estudiados previamente, ya que esta patología es sospechada por alteraciones en el hemograma que es solicitado con frecuencia ante cualquier estudio médico y que en general es una entidad más conocida en nuestro medio y por tanto más fácilmente sospechada. Por todos estos motivos, cabría esperar una prevalencia mayor de hemoglobinopatías en nuestra población.
Muchos de estos pacientes (53%) han conocido su alteración genética al realizarse el control rutinario de su diabetes o bien a partir de los hallazgos en el hemograma solicitado por otro motivo (20%). Por tanto, creemos que con esta sistemática de trabajo se está ayudando, en parte, a ampliar estudios en familiares y de esta manera proporcionar el diagnóstico temprano de una hemoglobinopatía en la descendencia, en estado homocigoto o heterocigoto doble, pudiendo así establecer las medidas oportunas para disminuir la mortalidad y morbilidad precoz de estas enfermedades (16). La baja proporción de familiares de primer grado encontrada en nuestros pacientes (16,6%; 15 núcleos familiares distintos) nos hace reflexionar sobre la necesidad de continuar insistiendo en la importancia que tiene el ampliar el estudio familiar, sobre todo en una población como la nuestra carente por el momento de sistemas de detección precoz de estas enfermedades.
En cualquier caso, este trabajo podría aproximarnos a la verdadera incidencia de las hemoglobinopatías en nuestros pacientes y puede ser el punto de partida para diseñar otros estudios sobre grupos poblacionales concretos, por ejemplo neonatos, adolescentes o gestantes y plantear el desarrollo de programas de control y prevención en la provincia de Santa Cruz de Tenerife. Probablemente en estos grupos poblacionales se encuentren aún más casos de hemoglobinopatías al incluir una mayor proporción de población inmigrante y por tanto, mayores tasas teóricas de estas enfermedades.
Bibliografía
1. Weatherall DJ, Clegg JB. The thalassaemia syndromes. 3ª ed. Oxford: Blackwell; 1981. [ Links ]
2. Drepanocitosis y otras hemoglobinopatías. Nota descriptiva nº 308. Organización Mundial de la Salud. Agosto 2006. [Acceso el 15 de Julio de 2007]. Disponible en: http://www.who.int/mediacentre/factsheets/fs308/es/index.html [ Links ]
3. Vives Corrons JL. Anemias por defectos congénitos de la hemoglobina. Hemoglobinopatías estructurales y talasemias. Medicine 2001; 8: 2684-93. [ Links ]
4. Dulín Íñiguez E, Cantalejo López MA, Cela de Julián ME, Galaron García P. Detección precoz neonatal de anemia falciforme y otras hemoglobinopatías en la Comunidad Autónoma de Madrid. Estudio piloto. An Pediatr (Barc) 2003; 58: 146-55. [ Links ]
5. Cabot Dalmau, Casado Toda M, Barberán Pérez J, Roqueta Sureda M, Martorell Aymerich Q, Bosch Llobet A, et al. Screening neonatal de drepanocitosis en el Consorci Sanitari de Mataró. Justificación y primeros resultados. An Esp Pediatr 1998; 49: 157-60. [ Links ]
6. Malcorra Aspiazu JJ. Hemoglobinopatías estructurales en Canarias. Haematologica. 1998; 83 (Supl. 1): 413-9. [ Links ]
7. Calvo-Villas JM, Zapata Ramos MF, Cuesta Tovar J, De la Iglesia Iñigo S, Ropero Gradilla P, et al. Prevalencia de hemoglobinopatías en mujeres gestantes en el área sanitaria de Lanzarote. An Med Interna (Madrid) 2006; 23: 206-12. [ Links ]
8. Baiget M. Hemoglobinopatías estructurales en España. Sangre (Barc) 1985; 30: 899-904. [ Links ]
9. Pérez Sirvent M, Moreno Miralles I, Bolufer Gilabert P. Alteraciones moleculares de las talasemias en España. Revisión de los estudios existentes. Med Clin (Barc) 1999; 113: 789-94. [ Links ]
10. Malcorra JJ, Balda MI, Campo C, Mataix R, Molero T, Castro E. Hemoglobinopatías y talasemias en la colonia hindú afincada en las Islas Canarias. Sangre 1993; 38: 342. [ Links ]
11. Oliva Berini E, Cladera Serra A, Torrent Quetglas M. Campaña para la detección de la betatalasemia minor y prevención de la major en la isla de Menorca. Experiencia de diez años. Campaña para la detección de la betatalasemia minor y prevención de la major en la isla de Menorca. Experiencia de diez años. Med Clin (Barc) 1998; 110: 361-4. [ Links ]
12. Memoria de actividades 2005. Hospital Universitario "Nuestra Señora de Candelaria" de Santa Cruz de Tenerife. [ Links ]
13. Cela de Julián E, Dulín Íñiguez E, Guerrero Soler M, Arranz Leirado M, Galarón García P, Meléndez Bieler C, et al. Evaluación en el tercer año de implantación del cribado neonatal universal de anemia falciforme en la Comunidad de Madrid. An Pediatr (Barc) 2007; 66 (4): 382-6. [ Links ]
14. Dirección General de Salud Pública de Extremadura. Programa de detección precoz de minusvalías, 2003. [ Links ]
15. Mañú Pereira MM, Maya A, Cararach V, Sabriá J, Boixareda J, Quintó L, et al. Cribado neonatal de hemoglobinopatías y déficit de glucosa-6-fosfato deshidrogenasa en Cataluña. Estudio piloto en población anómima no relacionada. Med Clin (Barc) 2006; 126: 281-5. [ Links ]
16. Ruano Raviña A, Jato Díaz M. Cribado neonatal de hemoglobinopatías. Santiago de Compostela: Servicio Galego de Saúde, Axencia de Avalación de Tecnoloxias Sanitarias de Galicia, avalia-t; 2004. Serie Avaliación de tecnologías. Informes de evaluación INF2004/004. [ Links ]
17. Instituto Nacional de Estadística. Nota de prensa del 1 de marzo de 2007 referida a 1 de enero de 2006. [Acceso el 10 de Julio de 2007]. Disponible en: www.ine.es/prensa/prensa.htm [ Links ]
18. Datos del ISTAC (Instituto de Estadística de Canarias) referidos a 2006. [Acceso el 5 de Junio de 2007]. Disponible en: http://www.gobcan.es/ istac/estadisticas.html. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Silvia de las Heras Flórez.
Hospital Universitario Nuestra Señora de Candelaria.
Ctra. del Rosario, 145.
Santa Cruz de Tenerife.
Trabajo aceptado: 15 de octubre de 2007

