










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkAvances en Odontoestomatología
versão On-line ISSN 2340-3152versão impressa ISSN 0213-1285
Av Odontoestomatol vol.21 no.6 Madrid Nov./Dez. 2005
Bases moleculares del cáncer oral. Revisión bibliográfica
García García V*,González-Moles MA**, Bascones Martínez A***
|
INTRODUCCIÓN
El cáncer supone un problema de salud a nivel mundial. Es una de las tres causas más importantes de mortalidad humana (Cáncer-Corazón-Carretera). Una enfermedad intrigante, muchas veces inesperada, una sombra que oscurece la vida de miles de personas.
El cáncer oral representa del 2 al 4% de todos los cánceres diagnosticados, con un incremento anual de 5.000 nuevos casos por año 8 (1), aunque cabe destacar la marcada variabilidad geográfica respecto a su incidencia. Las cifras más altas las encontramos en la India y otras regiones del Sudeste asiático (representando el cáncer oral el 40% de todos los cánceres orgánicos), muy relacionadas con las diferentes formas de hábito tabáquico en dichas zonas. Se habla de la aparición de 56.000 casos nuevos cada año, esto supone que en un momento dado 100.000 personas se encuentren padeciendo la enfermedad (2,3,4). En Europa, Hungría destaca con una incidencia de cáncer oral del 5% respecto al 1-4% en el resto de los países europeos(5). La prevalencia del cáncer oral en España es de 11,4-17,4 /100.000 habitantes, siendo las localizaciones intraorales más frecuentes la lengua (25,1%) y el suelo de la boca (10,2%)(6,7).
El cáncer de la cavidad oral comprende áreas malignas localizadas en el labio, la lengua, el suelo de la boca, la encía, el paladar, la mucosa bucal / vestibular y las glándulas salivares(8). La manifestación clínica más frecuente de un cáncer incipiente es un área roja, pero también pueden aparecer pequeñas ulceraciones o zonas granulares. La enfermedad afecta predominantemente a personas mayores, en su gran mayoría del sexo masculino, a partir de los 40 años con un pico máximo a la edad de los 60 años(9).
Histológicamente, el carcinoma de células escamosas o epidermoide es el tumor más común encontrado en la cavidad oral constituyendo, aproximadamente, el 5% de todas las neoplasmas(9) y el 30% de los cánceres de cabeza y cuello(9,10). Según las incidencias recogidas en la base de datos de la Organización Mundial de la Salud en el año 2000, el carcinoma epidermoide es el octavo cáncer más común en todo el mundo(11).
Aunque la cavidad oral es un área accesible y examinada con frecuencia, es muy habitual el diagnóstico tardío del carcinoma epidermoide, asociándose por este motivo un porcentaje de supervivencia de tan sólo el 25% a los cinco años(12,1).
El cáncer oral es normalmente indoloro, pero puede diagnosticarse mediante un cambio de color rojizo o rojo-blanquecino en la mucosa, la aparición de una zona indurada, hemorragias sin causa aparente, presencia de una zona ulcerada crónica y existencia de una masa sobreelevada (verrucosa o ulcerada)(13). Nos obstante, hay algunos tipos de cánceres difícilmente detectables por el procedimiento clínico habitual. Tal es el caso del melanoma amelanótico maligno, el cuál, debido a su falta de pigmentación, la mayoría de las veces pasa desapercibido(14).
Es indiscutible el papel del odontólogo en la prevención primaria (promocionando un estilo de vida saludable) y secundaria (diagnóstico precoz) del cáncer oral, haciéndose imprescindible al tener en cuenta que en aproximadamente la mitad de los casos el cáncer oral va precedido por lesiones o condiciones precancerizables, por lo que un diagnóstico precoz de las mismas influirá notablemente en la tasa de supervivencia, reduciendo la morbilidad y mortalidad (6,8). Sin embargo, un gran porcentaje de tumores son diagnosticados en un estado de evolución muy avanzado conduciendo a la necesidad de tener que aplicar técnicas terapéuticas agresivas que alteran notablemente la calidad de vida de los enfermos(15). Entre estos procedimientos destaca la radioterapia, que al actuar no sólo en los tejidos enfermos, sino también sobre las células sanas, provoca la aparición de alteraciones orales (mucositis, xerostomía) y sistémicas (inmunosupresión, infecciones víricas y fúngicas), entre otras(9). Con el uso de la quimioterapia también se producen complicaciones a nivel de la mucosa gastrointestinal y oral mediante un mecanismo de toxicidad directa e indirecta. El mecanismo directo se explica porque las células de la mucosa oral presentan una alta tasa de proliferación celular semejante a las células neoplásicas, haciéndolas susceptibles a las diferentes modalidades de agentes citostáticos, dando como resultado atrofias y colapsos eventuales de la integridad de la mucosa debido al daño de la capa reproductiva y a la regeneración celular limitada. La toxicidad indirecta es el resultado de la inmunosupresión inducida por el tratamiento antineoplásico, de tal manera, que la cavidad oral está en riesgo de sufrir lesiones e infecciones mucosas y también puede tener lugar la exacerbación de lesiones preexistentes(16).
Entender las bases moleculares del cáncer oral supone una tarea ardua que trataremos de resolver simplificando lo máximo posible y ayudándonos de esquemas esclarecedores para comprender la raíz de esta inquietante y desconocida enfermedad.
Cáncer Oral
El carcinoma oral de células escamosas es un proceso multisecuencial que implica una serie de alteraciones discretas, irreversibles y complementarias en los genes que controlan el crecimiento, la muerte y la diferenciación celular(17).
Etiología: Factores de riesgo
La etiología del cáncer oral permanece desconocida. No obstante se sabe de la existencia de una serie de factores de riesgo que podrían actuar como agentes carcinógenos a la hora de producir el cáncer oral. En la siguiente tabla se resumen los principales factores de riesgo asociados al cáncer oral (Figura 1).

Estado precanceroso
El carcinoma oral de células escamosas se desarrolla siguiendo uno de estos dos caminos:
"De novo", desarrollándose directamente a partir de mucosa sana.
Siguiendo la secuencia: estado precanceroso (displasia epitelial)- carcinoma(18).
Aproximadamente un 50% de los carcinomas epidermoides se desarrollan sobre un epitelio previamente alterado. En el Simposium Internacional de Uppsala, Axéll T. y cols. definieron algunos conceptos básicos para entender la carcinogénesis oral. Entendemos por lesión precancerosa un tejido morfológicamente alterado en el cuál la aparición de cáncer es más probable que en su homólogo normal. Una condición precancerosa es un estado sistémico generalizado que predispone a un riesgo mayor de padecer cáncer(19).
En la siguiente tabla puede observarse la clasificación de las lesiones y condiciones precancerosas(20) (Figura 2).

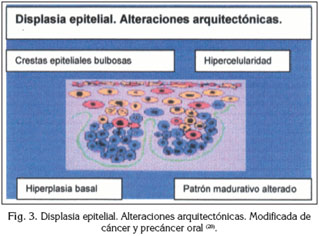
El potencial de malignización de estos estados llamados "potencialmente cancerosos", ya que la progresión hacia la malignidad no parece ser inevitable, viene determinado por la presencia de displasia epitelial, entendiéndose por esta la combinación variable de una serie de fenómenos microscópicos indicativos de un desorden de la maduración epitelial y una alteración de la proliferación celular(21,20). Los rasgos que identifican una displasia epitelial se clasifican en alteraciones arquitectónicas y alteraciones celulares (Figuras 3 y 4).

BASES MOLECULARES DEL CÁNCER ORAL
Carcinogénesis
La carcinogénesis es el mecanismo a través del cual se desarrolla una neoplasia maligna. Hay muchas lagunas en la literatura científica sobre este tema, si bien, poco a poco determinadas fases de la carcinogénesis van quedando esclarecidas.
La génesis y el desarrollo tumoral son el resultado de numerosas alteraciones moleculares que se producen en nuestro ADN y en donde están implicados los oncogenes, que a través de variaciones en su secuencia de nucleótidos adquieren capacidades generadoras de tumores. Los protooncogenes son secuencias génicas que codifican las proteínas que controlan el crecimiento y diferenciación celular. El resultado de la alteración de la secuencia génica del protooncogén, por mutación, amplificación o reordenamiento cromosómico, se conoce con el nombre de encogén(1).
La iniciación o activación del tumor empieza cuando el ADN de la célula o de una población de células se daña por la exposición a factores carcinogénicos, tanto endógenos como exógenos; la sensibilidad de las células mutadas respecto a su microambiente puede alterarse e iniciarse un crecimiento más rápido que en las células no alteradas(1).
La promoción tumoral se caracteriza por una expansión clonal selectiva de las células iniciantes, como resultado de la expresión alterada de los genes.
Los productos de dichas células se asocian a una hiperproliferación, inflamación y remodelación tisular(1).
La progresión del tumor se producirá cuando coincidan los factores implicados en la iniciación y promoción del mismo. Parece ser que el cáncer se desarrolla de forma gradual, a partir de células ligeramente aberrantes, y se caracteriza por una acumulación de alteraciones en los genes encargados de la regulación de la homeostasis celular, como los oncogenes, genes supresores de tumores y los genes reparadores de ADN(1).
Ciclo Celular
La acción de diversos oncogenes, aparece implicada en fenómenos de proliferación, diferenciación y supervivencia celular, así como en la capacidad de diseminación y metástasis de las células neoplásicas(22).
Los oncogenes codifican proteínas llamadas oncoproteínas, similares a los productos normales de los protooncogenes, salvo que las oncoproteínas carecen de algunos elementos reguladores importantes y su producción no depende de factores de crecimiento ni de otras señales externas(22).
Los factores de crecimiento estimulan la proliferación de las células normales y se cree que muchos de ellos intervienen en la tumorogénesis (mutaciones). La proliferación celular puede seguir los siguientes pasos:
Unión de un factor de crecimiento a su receptor específico existente en la membrana celular.
Activación del receptor del factor de crecimiento que, a su vez, activa a varias proteínas transductoras de señales existentes en la capa interna de la membrana plasmática.
Transmisión por el citosol de la señal transducida hasta que llega al núcleo. Inducción y activación de los factores reguladores de núcleos que inician la transcripción del ADN.
Paso de la célula al ciclo celular, por el que progresa hasta que se produce su división(22) (Figura 5).

Los receptores de crecimiento son proteínas transmembrana que sufren una activación transitoria cuando el receptor capta a su factor de crecimiento específico, a lo que sigue rápidamente la mitosis. Las versiones oncogénicas de estos receptores sufren activaciones persistentes sin necesidad de unirse al factor de crecimiento correspondiente. De esta forma, el receptor mutante libera hacia la célula continuas señales que estimulan la mitosis(22).
Recientemente, se ha intensificado el interés de los investigadores en la expresión de receptores de factores de crecimiento codificados por proto-oncogenes c-erb B en el desarrollo de cáncer oral. Uno de estos receptores es el receptor para el Factor de Crecimiento Epidérmico, que se une a ErbB-1, mientras que el factor de diferenciación neural (NDF) es el ligando preferente para ErbB-3 y ErbB-4. Estos receptores están involucrados en la proliferación y diferenciación celular, y su activación aberrante o su sobre-expresión puede contribuir al desarrollo y progresión tumoral (10).
Todas las rutas estimuladoras e inhibidoras del crecimiento y proliferación celular convergen en un complejo aparato molecular presente en el núcleo denominado reloj del ciclo celular. Dicho reloj se descontrola en prácticamente todos los tipos de cánceres humanos(20). En la figura 6 se pueden observar las distintas fases del ciclo celular.

La progresión ordenada de las células a través de las distintas fases del ciclo celular depende de las ciclinas, de las quinasas dependientes de ciclinas (CDK) y de sus inhibidores. Las mutaciones que alteran la regulación de la actividad de las ciclinas y de las CDK favorecen la proliferación celular(22).
Genes supresores del cáncer
Los genes supresores tumorales están implicados en diversos procesos de división celular: la regulación de la expresión génica, control del ciclo celular, programación de la muerte celular y estabilidad del genoma. La pérdida de actividad de estos genes provoca la incapacidad de respuesta a los mecanismos de control que regulan la división celular; de modo que se produce una proliferación más o menos incontrolada de la célula, lo cual conduce, en ocasiones, al desarrollo de neoplasias y a la evolución de las mismas hacia procesos tumorales más agresivos(23).
Gen Rb
El gen del retinoblastoma es el primer gen supresor de tumores conocido(24). Produce la pRB que actúa como un freno al progreso de la célula desde la fase G1 a la fase S del ciclo celular. Cuando la célula recibe el estímulo de los factores de crecimiento, la proteína Rb se inactiva, por lo que el freno desaparece y la célula atraviesa el punto de control G1-S. Si falta la proteína Rb o si una mutación altera su capacidad, el freno molecular del ciclo celular desaparecerá y la célula avanzará despreocupadamente hacia la fase S(22).
La ciclina D1 es una proteína inestable fundamental para la progresión de G1 mediante la activación del complejo CDK4/6 que lleva a la fosforilación de la proteína pRB. La fosforilación de la proteína pRB es esencial para la continuación del ciclo celular. Si pRB no está fosforilada se bloquea el ciclo celular. Se ha encontrado sobreexpresión de Ciclina D en carcinomas orales(25,20,26). pRb fosforilada libera un factor de transcripción que induce la activación de los genes que regulan la síntesis de ciclina E y ciclina A:
La síntesis de ciclina E comienza en la fase G1 tardía. Esta ciclina se une a la CDK2, formando un complejo activo que induce la síntesis de las enzimas y proteínas necesarias para la producción de ADN.
La ciclina A tiene la misión de activar a la CDK1 durante la fase S. El complejo ciclina A/CDK1 promueve, durante la fase G2, la síntesis de las proteínas implicadas en la mitosis. Estas proteínas conducen finalmente a la división de la célula en la fase M (4).
Las CDK también son reguladas por los llamados inhibidores de las CDK (CDKI), de los cuales encontramos dos familias en mamíferos: la familia de p21 (p21Cip1/ WAF1), p27Kip1, p57Kip2) y la familia INK4 (p15INK4b, p16INK4a, p18INK4c,p19INK4d)(25).
La p16 se une al complejo CDK4/CDK6 impidiendo su asociación con la ciclina D1 dando como resultado la detención del ciclo celular en G1 mediante la no fosforilación de pRB(27).
El CDKI p27Kip1 comparte secuencias homólogas con p21 y actúa como un regulador negativo del ciclo celular implicado en la detención en la fase G1 mediante un mecanismo independiente de p53(25).
Gen p53
Este gen, en condiciones de normalidad, codifica una fosfoproteína nuclear (proteína natural o salvaje) que actúa como un regulador negativo de la proliferación celular mediante una acción compleja, puesto que al mismo tiempo actúa como factor de transcripción, interruptor del ciclo celular e inductor de apoptosis. Cuando ocurren mutaciones en p53, se produce una síntesis anormal de proteína, proteína p53 mutada, que tiende a estabilizarse y acumularse en el núcleo. Esta proteína mutada pierde su capacidad supresora del crecimiento celular. La regulación negativa del p53 sobre la replicación ocurre ante diferentes tipos de agresiones en el ADN, aumentando entonces la cantidad de proteína nuclear p53. La consecuencia inmediata del incremento de p53 es la detención del ciclo celular en G1, actuando mientras los mecanismos de reparación del ADN(24).
Si la reparación del ADN es satisfactoria, p53, activará a un gen denominado mdm2, cuyo producto se une e inhibe a la propia p53, levantando así el bloqueo celular22.Si las alteraciones del ADN son muy extensas y el daño no puede ser reparado, la proteína p53 puede inducir el inicio de muerte celular fisiológica (apoptosis). De esta forma, la proteína salvaje p53 actúa como un verdadero guardián de la integridad del genoma, estableciendo la posibilidad de que las células dañadas reparen su ADN, previniendo la inestabilidad genómica.
La p53 bloquea el ciclo celular mediante la inducción de la proteína p21, inhibidora de las CDK. La p21 bloquea la transición G1-S y bloquea también directamente la replicación del ADN en la fase S del ciclo celular mediante la inhibición de la actividad de la proteína PCNA sobre la ADN polimerasa d27. También se ha visto como la p21 puede actuar por otro mecanismo independiente de la p53(25,20).
El gen p53 bloquea también la angiogénesis, posible favorecedora del desarrollo y la diseminación tumoral, a través de la secreción de trombospondina-1 (TSP-1) por parte de los fibroblastos; de esta forma, en las células transformadas, la ausencia de p53 favorecería la inducción de la angiogénesis mediante una disminución de la secreción de TSP-1(24).
Gen p73
En condiciones adecuadas, de igual manera que el gen p53, puede detener el ciclo celular y provocar la apoptosis(22).
Genes BRCA-1 Y BRCA-2
Son 2 genes supresores del cáncer. Ni el BRCA-1 ni el BRCA-2 se encuentran inactivados en la mayoría de los carcinomas de células escamosas, por lo que se cree, que existen nuevos genes supresores tumorales implicados(22). Tal es el caso del gen supresor tumoral FHIT que ha aparecido recientemente alterado en algunas líneas celulares de carcinomas de cabeza y cuello(27), aunque todavía no está muy claro este papel y se necesitan más estudios para corroborarlo(28).
Apoptosis
Existen básicamente dos formas de muerte celular: apoptosis y necrosis. Apoptosis es un término griego que hace referencia a la caída de los pétalos de las flores o de las hojas de los árboles. El proceso de apoptosis tarda aproximadamente 30 minutos. Los fenómenos que se producen son: la rotura del citoesqueleto y las organelas, el encogimiento celular, brote de yemas en la membrana celular, condensación de la cromatina y formación de cuerpos apoptóticos que son fagocitados por los macrófagos o las células vecinas. La necrosis, al contrario que la apoptosis, provoca una respuesta inflamatoria debido a que la célula se hincha provocando la rotura de la membrana celular y liberando al exterior todo su contenido(29).
La apoptosis es un proceso activo, genéticamente controlado y destinado a la autoeliminación celular. Es un fenómeno imprescindible para mantener la homeostasis, ya que el crecimiento celular mediante la proliferación celular debe ser equivalente a la degradación tisular mediante muerte celular o apoptosis(30). Distinguimos 3 fases en el proceso apoptótico: iniciación, ejecución y degradación. La fase de iniciación es activada por una gran variedad de señales que incluyen el daño del ADN, hipoxia, falta de nutrientes, factores de crecimiento u hormonas y la activación de los receptores de la muerte. Las señales que estimulan las dos fases siguientes son menos variadas. La apoptosis es regulada por un gran número de moléculas como la familia bcl-2, el factor de necrosis tumoral (TNF), genes supresores de tumores, oncogenes, factores mitocondriales y la familia de las caspasas. Por el momento se conocen dos mecanismos para llevar a cabo la apoptosis:
Mecanismo extrínseco: activación transmembrana de los receptores de muerte de la superfamilia del TNF que lleva a la activación de la caspasa –8. La unión del TNF-α al receptor Fas (miembro de la familia de los receptores del TNF) conduce al reclutamiento de la forma zimógena de la caspasa –8 para formar un complejo llamado "complejo de inducción de la señal de muerte" (DISC). DISC activa la caspasa –8 comenzando la cadena de proteólisis de las caspasas 3, 6, 7 y terminando en la apoptosis. Por otro lado la activación de Fas hace que la caspasa-8 provoque la escisión de Bid en tcBid, lo que inducirá cambios apoptogénicos mitocondriales.
Mecanismo intrínseco: inducido por el estrés celular, implicando a la mitocondria que libera factores apoptogénicos como el citocromo c, el factor inductor de la apoptosis (AIF), ATP, proteínas del sock calórico y Diablo/Smac. El citocromo c junto con el ATP y el factor activador de la proteasa apoptótica (Apaf-1) forman un complejo llamado apoptosoma, que regula la activación de la caspasa-9 que a su vez activará como en el mecanismo extrínseco a las caspasas 3,6 y 7(29) (Figura 7).

Junto con este complejo mecanismo de activación apoptótica hay que reseñar la existencia de moléculas inhibidoras de la apoptosis (IAPs) que son: NIAP, CIAP1, CIAP2 y la proteína apoptótica inhibidora del cromosoma X (XIAP)(63). También se ha visto como el Factor Nuclear kappa B aumenta el proceso cancerígeno mediante la disminución de la apoptosis mediada por TNF(64,65). La survivina es una IAP recientemente descubierta. Se encuentra sobreexpresada en gran número de neoplasias, pero suele ser indetectable en la mayoría de los tejidos sanos diferenciados(34).
La p53 elimina selectivamente las células estresadas o dañadas mediante apoptosis protegiendo al organismo del desarrollo de tumores. p53 puede mediar la apoptosis a partir de la regulación de bcl-2, de la superfamilia del receptor TNF y también de un modo independiente mediante la regulación de Fas o afectando al potencial de la membrana mitocondrial(29).
CONCLUSIONES
Es indiscutible el papel del odontólogo en la prevención y diagnóstico precoz del cáncer oral. Para poder llevar a cabo este papel tan importante debe conocer a fondo las bases moleculares del cáncer oral y poder así actuar desde el comienzo, aumentando los índices de supervivencia de los pacientes oncológicos.
BIBLIOGRAFÍA
1. Jané Salas E. y cols. Importancia de la dieta en la prevención del cáncer oral. Med Oral 2003;8:260-8. [ Links ]
2. Tanuja T. y cols. Expresión of Bcl-2 and Bax In Chewing Tobacco-Induced Oral Cancers and Oral Lesions from India. Pathology Oncology Research Vol 8, No 2, 109-14, 2002. [ Links ]
3. Ravi D. y cols. Apoptosis, angiogenesis and proliferation:trifunctional measure of tumour response to radiotherapy for oral cancer. Oral Oncology 37 (2001);164-71. [ Links ]
4. Agarval S., y cols. MDM2/p53 co-expression in oral premalignant and malignant lesions: potencial prognostic implications. Oral Oncology 35 (1999);209-16. [ Links ]
5. Bánóczy J., Rigó O: Prevalence study of oral precancerous lesions within a complex screening system In Hungary. Community Dent Oral Epidemiol 1991; 19: 265-7. [ Links ]
6. Seoane J. y cols. Assessment of dental studentsability to recognise precancerous lesions and conditions.Eur J Dent Educ 1997; 1: 172-5. [ Links ]
7. Martinez-Conde R. y cols. Factores clínico-patológicos en el carcinoma de células escamosas inicial de lengua y suelo de la boca, en Vizcaya. Medicina Oral 2001; 6: 87-94. [ Links ]
8. Silverman S. Demographics and occurrence of oral and pharyngeal cancers.JADA, Vol. 132, November 2001. [ Links ]
9. Caribé Gomes F. y cols. Manejo odontológico de las complicaciones de la radioterapia y quimioterapia en el cáncer oral. Med Oral 2003; 8: 178-87. [ Links ]
10. De Vicente J.C. y cols. Expresión de las proteínas de los proto-oncogenes ErbB-3 y ErbB-4 en el carcinoma oral de células escamosas: estudio piloto. Med Oral 2003; 8: 374-81. [ Links ]
11. Tsai W. y cols. The mRNA profile of genes in betel quid chewing oral cancer patients. Oral Oncology (2004) 40 418-26. [ Links ]
12. Gonzalez – Moles M.A. y cols. P21WAF1/CIP1 Protein and Tongue Cancer Prognosis. Anticancer Research 24: 3225-32 (2004). [ Links ]
13. Bouquet J.E./ Whitaker S.B. Oral leukoplakia- Rationale for diagnosis and prognosis of its clinical subtypes or "phases". Quintessence Int 1994; 25: 133-40. [ Links ]
14. Notani K. y cols. Amelanotic malignant melanomas of the oral mucosa. British Journal of Oral and Maxillofacial Surgery (2002) 40, 195-200. [ Links ]
15. Gonzalez-Moles M.A. y cols. Prognosis value of the expresión of Ki-67 for squamous cell carcinoma of the oral cavity. Acta Stomatologica Belgica, Anno 93, nº4, 1996. [ Links ]
16. Rojas de Morales T. Prevención oral en niños con cáncer: efectividad de un protocolo. Medicina Oral 2001; 6: 326-34. [ Links ]
17. Contreras Vidaurre E.G. y cols. Retinoides: su aplicación en las lesiones precancerosas y el cáncer oral. Medicina Oral 2001; 6: 114-23. [ Links ]
18. Okazaki Y. y cols. Investigation of environmental factors for diagnosing malignant potencial in oral epithelial dysplasia. Oral Oncology 38 (2002) 562-73. [ Links ]
19. Axéll T. y cols. Oral white lesions with special referente to precancerous and tobacco-related lesions: conclusions of an internacional symposium held in Uppsala, Sweden, May 18-21 1994. J.Oral Pathol Med 1996; 25:49-54. [ Links ]
20. Bascones A. y cols. Cáncer y Precáncer Oral. 1ª Edición. Ediciones Avances. Madrid 2003. [ Links ]
21. Mc Alinden RL. y cols. Bcl-2 expression in sequential biopsies of potentially malignant oral mucosal lesions assessed by immunocytochemistry. Oral Diseases (2000) 6, 318-26. [ Links ]
22. Saiz Rodríguez A. Bases moleculares del cáncer oral. Med Oral 2001; 6:342-9. [ Links ]
23. López –Martínez M. y cols. Aplicaciones clínicas del diagnóstico de las alteraciones de p53 en el carcinoma escamoso de cabeza y cuello. Med Oral 2002; 7: 108-20. [ Links ]
24. Vera Sempere F.J., Navarro Hervas M. Sobreexpresión del gen supresor p53 en el cáncer oral. Medicina Oral 1997; 2: 283-96. [ Links ]
25. Schoelch M. y cols. Cell cycle proteins and the development of oral squamous cell carcinoma. Oral Oncology 35 (1999) :333-42. [ Links ]
26. Kudo Y. y cols. p27Kip1 Accumulation by Inhibition of Proteasome Function Induces Apoptosis in Oral Squamous Cell Carcinoma Cells. Clinical Cancer Research Vol. 6, 916-923, March 2000. [ Links ]
27. Jefferies S.,Foulkes W.D. Genetic mechanisms in squamous cell carcinoma of the head and neck. Oral Oncology 37 (2001) 115-26. [ Links ]
28. Pateromichelakis S. y cols. The FHIT gene in oral squamous cell carcinoma: allelic imbalance in frequent but cDNA aberrations are uncommon. Oral Oncology 36 (2000) 180-8. [ Links ]
29. Loro L. y cols. Cell death regulation in oral squamous cell carcinoma: methodological considerations and clinical significance. J Oral Pathol Med 2003;32: 125-38. [ Links ]
30. Petrasch S. y cols. Neoadjuvant hyperfracionated irradiation induces apoptosis and decreases proliferation in squamous cell cancer of the oral cavity. Int J Oral Maxillofac. Surg. 2000;29: 285-9. [ Links ]
31. Matsumiya T. y cols. Cisplatin inhibits the expression of X-chromosome-linked inhibitor of apoptosis protein in an oral carcinoma cell line. Oral Oncology 37(2001) 296-300. [ Links ]
32. Azuma M. y cols. Cisplatin induces apoptosis in oral squamous carcinomacells by the mitochondria-mediated but not the NF-kB-suppressed path way. Oral Oncology 39 (2003) 282-9. [ Links ]
33. Chen S. y cols. Potentiation of Tumor Necrosis Factor-mediated Apoptosis of Oral Squamous Cell Carcinoma Cells by Adenovirus-Mediated Gene Transfer of NF-kB Inhibitor. J Dent Res 2002; 81 (2): 98-102. [ Links ]
34. Muzio L. y cols Survivin, a Potencial Early Predictor of Tumor Progresión in the Oral Mucosa. J Dent Res 2003; 82(11): 923-8. [ Links ]
CORRESPONDENCIA
Virginia García García
virginiaborj@hotmail.com
639910423.

