










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.78 no.2 feb. 2003
COMUNICACIÓN CORTA
LA MUTACIÓN P28T DEL GEN GK1 COMO CAUSA DE
UNA DEFICIENCIA FAMILIAR DE GALACTOQUINASA
MUTATION P28T IN GENE GK1 AS THE CAUSE OF A FAMILIAL
GALACTOKINASE DEFICIENCY
GIRÓS M1, BÓVEDA MD2, VÁZQUEZ DE LA CRUZ A3, LÁZARO P3, GATA A4,
SOLAR BOGA A3, BRIONES P5
| RESUMEN Objetivo/Método: Alertar sobre la deficiencia de galactoquinasa (GK), como posible causa de cataratas infantiles, e incluso cataratas preseniles en los heterozigotes portadores. El diagnóstico mediante determinación del enzima y del galactitol, permitiría la introducción de una dieta exenta de galactosa que previene totalmente el daño. Palabras clave: Galactosemia, cataratas, etnia gitana. | SUMMARY Objective/Method: To alert about galactokinase deficiency (GK) as a possible cause of infantile cataracts, and even presenile cataracts in heterozygous carriers. Diagnosis by enzyme and galactitol determination would lead to the introduction of a galactose-free diet which completely prevents the damage. Key words: Galactosemia, cataracts, gypsy ethnia. |
Recibido: 20/2/02. Aceptado: 17/7/02.
Institut de Bioquímica Clínica. Corporació Sanitaria. Barcelona. España.
1 Doctora en Ciencias Químicas.
2 Licenciada en Ciencias Químicas. Hospital Clínico Universitario. Santiago.
3 Licenciado en Medicina. Hospital Infantil Juan Canalejo. A Coruña.
4 Licenciada en Biología.
5 Doctora en Ciencias Biológicas. CSIC. Barcelona.
Comunicación presentada parcialmente como póster al IV Congreso Nacional de Errores Congénitos del Metabolismo (Tenerife 2001).
Los autores manifiestan no tener interés comercial ni haber recibido apoyo económico para la realización del mismo.
Correspondencia:
Marisa Girós
Institut de Bioquímica Clínica
Edificio Helios III planta baja
C/. Mejía Lequerica, s/n
08028 Barcelona
España
E-mail: mgiros@clinic.ub.es
INTRODUCCIÓN
La deficiencia de la galactoquinasa (GK) es un defecto congénito del metabolismo de la galactosa que, a diferencia de la galactosemia clásica, no presenta manifestaciones severas en el neonato. Su principal sintomatología clínica es el desarrollo de cataratas durante las primeras semanas o meses de vida, causadas por la acumulación de galactitol en el cristalino. Se ha descrito también la predisposición de los portadores de la deficiencia a desarrollar cataratas preseniles (20-50 años). En ambos casos el daño se puede prevenir totalmente mediante un diagnóstico adecuado y tratamiento con dieta exenta de galactosa (1,3).
Los resultados obtenidos en el despistaje neonatal para galactosemia en diferentes poblaciones caucásicas y en Japón, muestran que la deficiencia de GK es una condición rara (1/1.000.000), pero sin embargo, con una frecuencia especialmente elevada en los individuos de etnia gitana en Europa. Estudios realizados en 6 familias Romani de Bulgaria identificaron una misma mutación (P28T) en todos ellos, a la que se ha atribuido un efecto fundador (1).
Nosotros presentamos el caso de una familia española de etnia gitana con alto grado de consanguinidad, con tres hembras de fratrias distintas afectas de deficiencia de GK, homocigotas para la mutación P28T en el gen GK1.
CASO CLÍNICO
Se trata de una familia española de etnia gitana con alto grado de consanguinidad. El estudio de despistaje metabólico para galactosemia resultó positivo en dos primas (caso 1 y caso 2) totalmente asintomáticas. En las niñas la actividad GK era indetectable y los niveles de galactitol incrementados (tablas I y II). El estudio de sus respectivos padres mostró actividades intermedias en tres de ellos, demostrando su carácter de heterozigotos; pero la madre del caso 2, afectada de ceguera, presentaba actividad GK indetectable y niveles de galactitol plasmático elevados (tablas I y II). El estudio molecular puso de manifiesto que la mutación P28T era la causante de la deficiencia en esta familia (fig. 1).
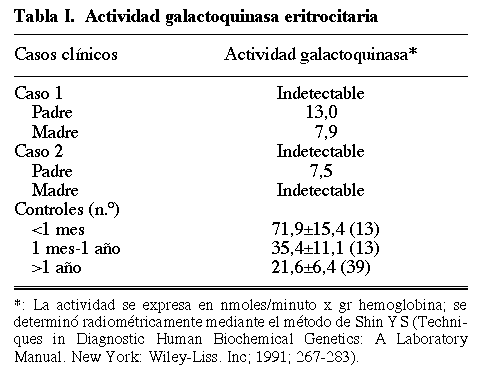

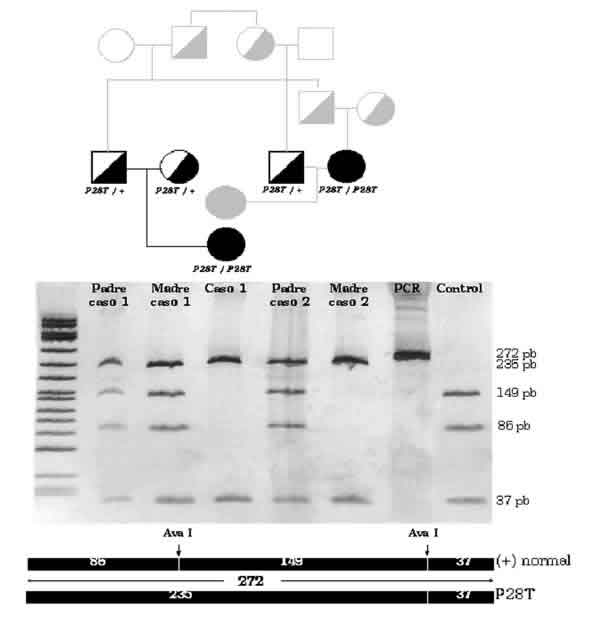
Fig. 1. Árbol genealógico e identificación de la mutación P28T mediante amplificación por PCR y análisis con enzima de restricción (AvaI) siguiendo lo descrito por Kalaydjeva y col (1999) (2). En el alelo normal (+) el fragmento amplificado por PCR [272 pares de bases (pb), carril "PCR"] se escinde en tres fragmentos de 37, 86 y 149 pb por digestión con el enzima AvaI (ver carril "control"). P28T causa la pérdida de una diana de restricción y en los individuos afectos sólo aparecen dos fragmentos de 37 y 235 pb (carriles "Caso 1" y "Madre caso 2"). En los heterotizogtes coexisten los fragmentos de 37, 86, 149 y 235 pb (carriles "Padre y Madre caso 1" y "Padre caso 2").
DISCUSIÓN
En el momento del diagnóstico cabe destacar los elevados niveles de galactitol en el caso 2, hija de homocigota afecta, respecto de los de su prima, hija de una portadora (tabla II). Sin embargo, ninguna de ambas había desarrollado cataratas. Estos resultados corroboran previas observaciones de que el galactitol atraviesa la barrera placentaria sin aparentes efectos adversos al feto y en el recién nacido (2).
Tras la introducción de dieta exenta de galactosa los niveles de galactitol fueron bajando, situándose por debajo de los 20 µmoles/L. Éste es el nivel de valores que se suele considerar aceptable como indicativo de buen cumplimiento de la dieta en los individuos afectos de galactosemia clásica (3).
Por otra parte, los estudios genéticos mostraron que las tres hembras afectas son homocigotas para la mutación P28T del gen GK1. Este gen está localizado en la región 17q24 y comprende 8 exones que codifican una proteína de 392 aminoácidos. Se han descrito unas 20 mutaciones distintas, la mayoría confinadas a una sola familia (4). La mutación P28T está producida por una transversión CA en el nucleótido 563 según la numeración del GenBank (n.º acceso L76927), que causa un cambio de una prolina por una treonina en el aminoácido en posición 28 (1). Este residuo de prolina está conservado desde bacterias a humanos y está cercano a la llamada secuencia de firma de la galactokinasa, que es una de las tres regiones muy conservadas de este gen (1,4). Esta mutación P28T ha sido descrita como prevalente en los individuos GK-deficientes de etnia gitana Romani de Bulgaria y se le ha atribuido un efecto fundador en esta población, donde la frecuencia de portadores es del 5% (1). Estudios realizados por nuestro grupo sugieren que esta frecuencia es también elevada, aproximadamente 2%, en los gitanos españoles (5).
Dado que la deficiencia de galactoquinasa puede ser una causa frecuente de ceguera en los niños y adultos de etnia gitana, es importante tener en cuenta esta condición en el diagnóstico diferencial de cataratas infantiles o adultas precoces, especialmente en individuos de dicho grupo de población.
AGRADECIMIENTOS
A la Dra. Laura Gort por sus sugerencias y discusión en el estudio mutacional.
A Avelina Vega y Sonia Moliner por su colaboración técnica en el trabajo.
BIBLIOGRAFÍA
1. Kalaydjieva L, Pérez-Lezaun A, Angelicheva D, Onengut S, Dye D, Bosshard UN et al. A founder mutation in the GK1 gene is responsible for galactokinase deficiency in Roma (Gypsies). Am J Hum Genet 1999; 65: 1299-1307. [ Links ]
2. Briones P, Girós M, Martínez V. Second spontaneous pregnancy in a galactosaemic woman homozygous for the Q188R mutation. J Inherit Metab Dis 2001; 24: 79-80. [ Links ]
3. Baldellou A, Baraibar R, Briones P, Ruiz M. Protocolo para el diagnóstico y el tratamiento de los errores congénitos del metabolismo de la galactosa. An Esp Pediatr 2000; 53: 1-9. [ Links ]
4. Hunter M, Angelicheva D, Levy HL, Pueschel SM, Kalaydjieva L. Novel mutations in the GALK1Gene in patients with Galactokinase Deficiency. Hum Mut Mutation in Brief 394 (2000) Online. [ Links ]
5. Hunter M, Heyer E, Austerlitz F, Angelicheva D, Nedkova V, Briones P et al. The P28T mutation in the GALK1 gene accounts for galactokinase deficiency in Roma (Gypsy) patients across Europe. Pediatr Res 2002; 51: 602-606. [ Links ]

