










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.78 no.12 dic. 2003
COMUNICACIÓN CORTA
FUNDUS ALBIPUNCTATUS ASOCIADO A DISTROFIA DE CONOS.
A PROPÓSITO DE UN CASO FAMILIAR
FUNDUS ALBIPUNCTATUS ASSOCIATED WITH CONE DYSTROPHY.
A FAMILIAL CASE REPORT
DÍAZ RODRÍGUEZ E1, CABALLERO ROMERA A2
| RESUMEN Se presenta un caso familiar de fundus albipunctatus asociado a distrofia de conos. Palabras clave: Fundus albipunctatus, distrofia de conos, 11-cis retinol deshidrogenasa. | SUMMARY We report a familial case of fundus albipunctatus associated with cone dystrophy. Key words: Fundus albipunctatus, cone dystrophy, 11-cis retinol dehydrogenase. |
Recibido: 19/5/03. Aceptado: 24/11/03.
Hospital General Juan Ramón Jiménez. Huelva. España.
1 Licenciado en Medicina. Servicio de Oftalmología.
2 Licenciado en Medicina. Servicio de Neurofisiología clínica.
Correspondencia:
Eusebio Díaz Rodríguez
C/. Dámaso Alonso, 9, Bellavista
21122 Huelva
España
E-mail: ejdr28@terra.es
INTRODUCCIÓN
Se aporta un caso familiar de fundus albipunctatus (FA) asociado a distrofia de conos (DC).
CASO CLÍNICO
Caso índice
Varón de 38 años diagnosticado en otro centro de retinitis pigmentosa que acude a revisión por fotodisforia, alteración en la visión de los colores y empeoramiento de la visión nocturna (sobre todo en ambientes con predominio de luz artificial) en los últimos meses.
Presenta una visión de 0,7 en ambos ojos; en el fondo de ojo destaca la presencia por toda la retina ecuatorial y posterior de depósitos amarillentos bien definidos y de distribución homogénea localizados en epitelio pigmentario (EPR)-retina externa asociados a pérdida de EPR en algunas zonas; llama la atención una coloración anaranjada paramacular que sugiere atrofia del EPR aunque sin espiculación ni agrupación del mismo. Los vasos retinianos y papilas son normales (fig. 1a).

los típicos depósitos del FA; tanto en el área macular
como en la retina ecuatorial que se visualiza se observan
áreas de atrofia del EPR.
En la angiografía con fluoresceína (AFG) se visualiza una hiperfluorescencia difusa que en el área paramacular adopta un patrón en anillo atípico por su tamaño y asociada a una pérdida fundamentalmente del EPR (fig. 1b).

Hiperfluorescencia difusa asociada a pérdida del EPR;
maculopatía en anillo.
En la campimetría se aprecia un escotoma central sin constricción del campo periférico (fig. 1c).
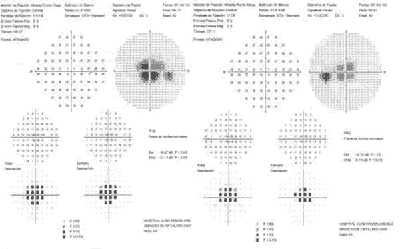
Segundo caso
Varón de 32 años, hermano del anterior, que acude a revisión. Refiere hemeralopia desde que tiene conocimiento y, en los últimos tiempos, ligera fotodisforia y leve disminución de la agudeza visual. La visión es de 1,0 en ambos ojos; tanto el fondo de ojo como la angiografía son similares a las de su hermano, aunque con menores alteraciones maculares y del EPR (figs. 2a y 2b). En la campimetría aparece un estotoma central (fig. 2c).

Numerosos depósitos amarillentos localizados por
mácula y retina ecuatorial (más abundantes que en el
primer caso). No se aprecia atrofia perimacular.

con tinción de los depósitos; inicio de maculopatía
en anillo de las mismas características que las de su hermano.

Se realizan sendos estudios electrofisiológicos no estandarizados consistentes en un primer electrorretinograma (ERG) basal y un segundo realizado a los 180 minutos de adaptación a la oscuridad. En ambos casos, el ERG basal revela una disminución evidente de la amplitud de todas las ondas. Tras la adaptación a la oscuridad, se pone de manifiesto en el primer caso una tendencia a la normalización del flash y escotópico, no modificándose ni el flicker ni el fotópico. En el segundo caso, la adaptación a la oscuridad normaliza las ondas, incluido el estudio de conos que se queda en el límite inferior de la normalidad para su edad.
DISCUSIÓN
El FA forma parte de las enfermedades conocidas en la literatura clásica con el nombre de Ceguera Congénita Nocturna Estacionaria. Se describe como una enfermedad autosómica recesiva con el único síntoma de ceguera nocturna «desde que el paciente tiene recuerdo» y unas alteraciones funduscópicas y electrofisiológicas muy características: estabilidad temporal, campimetría normal, recuperación del ERG tras la adaptación a la oscuridad y ausencia de signos que sugieran deterioro progresivo retiniano (1).
El caso familiar que presentamos tiene características típicas del FA. Sin embargo también tiene datos clínicos, oftalmoscópicos y electrorretinográficos de DC, sobre todo en el caso índice: alteraciones en la percepción de los colores, escotoma central y fotodisforia de comienzo reciente. La función de conos no mejora tras la adaptación a la oscuridad. En la AFG se aprecia una maculopatía en anillo atípica. En el segundo caso se evidencian signos incipientes de alteración de conos aunque el ERG tienda a normalizarse tras la adaptación a la oscuridad.
El diagnóstico puede realizarse teniendo en cuenta las características clínicas, oftalmoscópicas y electrofisiológicas. En ciertos casos cabría la confusión con un fenotipo tipo retinitis punctata albescens de una retinosis pigmentaria (RP). No existen síntomas ni signos de RP (la ceguera nocturna no es progresiva) y el comportamiento del ERG no es propio de la misma. El aparente empeoramiento de la visión nocturna del caso índice, referido sobre todo al conducir y en ambientes de predominio de la luz artificial, parece relacionado con la «ceguera nocturna urbana» descrita en las fases iniciales de una distrofia de conos más que con síntomas de la RP (1).
Todos los casos descritos hasta ahora de FA se deben a defectos genéticos en el ácido desoxirribonucleico (ADN) que codifica la enzima 11-cis retinol deshidrogenasa (Online Mendelian Inheritance in Man *601617 retinol dehydrogenase 5; RDH5 en http://www.ncbi.nlm.nih.gov/), enzima microsomal del ciclo visual localizada fundamentalmente en el EPR y que cataliza la oxidación a aldehído del 11-cis retinol (2,3). Recientemente, Nakamura y cols (4), describen en la serie de casos seguidos durante más tiempo la asociación frecuente de FA con DC a partir de la cuarta década y sugieren que ciertas mutaciones en RDH5 pueden originar FA aislado o asociado a disfunción clínica o subclínica de conos, descartando así la asociación casual. No obstante, permanece sin aclarar el papel de estas anomalías en la función de la enzima y cómo dan lugar a los diferentes fenotipos de la enfermedad.
Se desconoce la proporción de pacientes con FA que puedan desarrollar DC (hay pocos estudios que recojan la historia natural de la enfermedad) (5) así como la existencia de factores ambientales, incluido el ambiente genético, que modifiquen la expresión fenotípica del fundus albipunctatus en un sujeto determinado (efectos ambientales, heterogeneidad alélica, genes modificadores). Si es obvio que es preciso reconsiderar la actitud ante esta enfermedad y entenderla como una patología retiniana que puede provocar un deterioro progresivo y grave de la visión, sobre todo central.
BIBLIOGRAFÍA
1. Heckenlively JR, Arden GB. Principles and Practice of Clinical Electrophysiology of Vision. St. Louis: Mosby-Year Book, Inc; 1991. [ Links ]
2. Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus and other forms of stationary night blindness: LVII Edwuard Jackson Memorial Lecture. Am J Ophthalmol 2000; 130: 547-563. [ Links ]
3. Yamamoto H, Simon A, Eriksson U, Harris E, Berson EL, Dryja TP. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet 1999; 22: 188-191. [ Links ]
4. Nakamura M, Hotta Y, Tanikawa A, Terakasi H, Miyake Y. A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Invest Ophthalmol Vis Sci 2000; 41: 3925-3932. [ Links ]
5. Marmor MF. Long-term follow-up of the physiologic abnormalities and fundus changes in fundus albipunctatus. Ophthalmology 1990; 97: 380-384. [ Links ]

