










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.81 no.8 ago. 2006
REVISIÓN
Manejo terapéutico de la queratopatía asociada a aniridia congénita
Congenital aniridia keratopathy treatment
López-García J.S.1, García-Lozano I.2, Rivas L.3, Martínez-Garchitorena J.1
Hospital Central de Cruz Roja. Madrid. España.
1 Doctor en Medicina.
2 Licenciada en Medicina. Instituto de Ciencias Visuales (INCIVI). Madrid.
3Doctor en Biología. Hospital Ramón y Cajal. Madrid
Dirección para correspondencia
RESUMEN
Objetivo: Revisar la patogenia de la queratopatía por aniridia y establecer un protocolo de tratamiento según su gravedad.
Métodos: Revisión bibliográfica y experiencia personal en el manejo de pacientes con queratopatía por aniridia.
Resultados: Las manifestaciones oculares de esta anomalía incluyen defectos a nivel corneal, glaucoma, subluxación del cristalino y cataratas, hipoplasia de iris, fóvea y nervio óptico, ambliopía y nistagmus. La queratopatía por aniridia ocurre en un 20% de los pacientes. La correcta expresión del gen PAX6 es necesaria para el normal desarrollo corneal y actividad de las células limbares así como la correcta migración y adhesión de las células epiteliales.
Conclusiones: El manejo de los trastornos oculares debidos a la deficiencia limbal asociada a la aniridia es complejo pero ha cambiado en los últimos años. El concepto de célula madre limbal y de su microambiente ha modificado la aproximación terapéutica de estos pacientes. El empleo del suero autólogo, trasplante de membrana amniótica, trasplante de limbo o trasplante de células limbares cultivadas han sido utilizados en el tratamiento de distintos trastornos de la superficie ocular.
Palabras clave: Aniridia, insuficiencia limbal, suero autólogo, superficie ocular, ojo seco, epitelio corneal.
ABSTRACT
Objective: To attempt to review the aniridia keratopathy pathogenesis and establish a treatment protocol according to the severity of the symptoms.
Methods: Personal experience in aniridic keratopathy management and a bibliography review.
Results: The ocular manifestations of this anomaly include defects of the cornea, glaucoma, lens subluxation, cataracts, hypoplasia of the iris, fovea and optic nerve, amblyopia and nystagmus. The keratopathy occurs in a 20% of patients with aniridia. The correct PAX6 expression is necessary for normal corneal development, limbal stem cell activity and correct corneal epithelial cell migration and adhesion.
Conclusions: The management of ocular surface diseases due to limbal stem cell deficiency in aniridia is complex but has changed in recent years, as an understanding of the limbal stem cells and their microenvironment has modified the therapeutic approach. The use of autologous serum eye drops, amniotic membrane transplantation, limbal transplantation or cultivated limbal cell transplantation have all been reported as a treatment for several ocular surface diseases (Arch Soc Esp Oftalmol 2006; 81: 435-444).
Key words: Aniridia, limbal deficiency, autologous serum, ocular surface, dry eye, corneal epithelium.
Introducción
La aniridia es una enfermedad poco frecuente (1:65.000-95.000) producida por una alteración bilateral en el desarrollo del ojo, sin predilección de sexo o raza conocida. Se trata de una anomalía, con base genética bien conocida, que puede aparecer de forma esporádica o familiar, mostrando un patrón de herencia autosómico dominante con expresión variable entre los miembros de la familia. El gen responsable de la enfermedad (PAX6) se localiza en el brazo corto del cromosoma 11 (1). Este gen se expresa de manera extensa en el desarrollo de las distintas estructuras oculares incluyendo la córnea, el cristalino, el ángulo camerular, el cuerpo ciliar y todas las capas de la retina. Se trata, por tanto, de un trastorno global del ojo en el que la hipoplasia iridiana, que da nombre a la enfermedad, no es más que el signo clínico más aparente. Las alteraciones en el segmento anterior incluyen: queratopatía por disfunción limbar, ojo seco, glaucoma, cataratas, subluxación de cristalino y diversas anomalías en el ángulo camerular. En el segmento posterior destacan la hipoplasia macular y del nervio óptico siendo, además, frecuente la asociación de estrabismo y nistagmo que empobrecen el pronóstico visual desde edades tempranas (2) (fig. 1 ).

Fig. 1. Queratopatía asociada a aniridia.
Se puede apreciar la neovascularización superficial y la fibrosis subepitelial.
La primera descripción de la enfermedad la realiza Barratta en 1818, asignándole el nombre de irideremia en relación con el signo clínico más aparente, siendo ya en el siglo XX cuando comienza a usarse la denominación actual.
En humanos las mutaciones en el gen PAX6 se han encontrado no sólo en pacientes con aniridia sino también en algunos casos de Anomalía de Peter. Las mutaciones pueden ser intragénicas o delecciones del cromosoma 11 p. No se ha encontrado correlación entre el sitio de mutación del gen PAX6 y las variaciones en la expresión del fenotipo de aniridia, de hecho, una amplia gama de fenotipos puede ocurrir como consecuencia de una misma mutación.
Entre las alteraciones extraoculares más importantes destaca el desarrollo, en casos esporádicos, de tumor de Wilms, frecuentemente como parte del síndrome WAGR (tumor de Wilms, aniridia, anormalidades genitourinarias y retraso mental). La aparición de estas alteraciones extraoculares en la aniridia es más común en aquellos casos de aparición esporádica. Cuando se realiza el diagnóstico de anirida en un niño, cuya familia no tiene antecedentes, es importante establecer por análisis cromosómico si la delección se extiende al dominio del gen que predispone al tumor de Wilms para instaurar un régimen de monitorización durante el tiempo de riesgo de aparición de este tumor que suele ser hasta los 8 años.
Aunque el fenotipo puede variar considerablemente, incluso entre miembros de una misma familia, el defecto visual aparece ya en la primera década de la vida con agudezas visuales que oscilan entre el 10-20% por la hipoplasia foveal combinada con la posible existencia de cataratas congénitas, nistagmo y ambliopía (3). Frente a la estabilidad de las alteraciones retinianas, el progresivo deterioro de la función visual se debe fundamentalmente al desarrollo de glaucoma (50-75%), cataratas y queratopatía.
La queratopatía asociada a la aniridia (QAA) afecta a un 20% de los pacientes, aunque se detectan alteraciones de la superficie ocular hasta en un 90% de los casos (3). Las manifestaciones pueden llegar a comenzar en la primera década de la vida con engrosamiento irregular del epitelio periférico, seguido de neovascularización superficial que avanza centrípetamente en el transcurso de los años hasta afectar a toda la superficie corneal. Los pacientes sufren erosiones recurrentes, úlceras y dolor crónico y, si no son tratados, acaban presentando opacidad corneal grave por fibrosis subepitelial y cicatrización estromal (fig. 2 ).

Fig. 2. Opacidad corneal y catarata en paciente con aniridia congénita.
Bases genéticas y mecanismos patogénicos en la queratopatía asociada a la aniridia
El gen responsable de la aniridia se denomina PAX6 y se encuentra contenido en el segmento cromosómico 11p13 (4-6). La mayor parte de los trabajos recientes sobre las bases genéticas de la aniridia congénita provienen de estudios en animales, particularmente en ratones. Los modelos con alteración en el gen PAX6 de carácter homocigótico dan lugar a ratones con ausencia completa de ojos y cavidades nasales que mueren al poco tiempo de nacer. El defecto heterocigótico del PAX6 conduce al modelo de ratón Sey (Small eye) con microftalmos y alteraciones oculares similares a las observadas en humanos con anirida congénita, por lo que se considera un buen modelo experimental de la enfermedad.
El PAX6 es un gen que se encuentra contenido en una región del ADN considerada de extraordinaria importancia filogenética, ya que se ha conservado en numerosas especies de vertebrados e invertebrados a lo largo del proceso evolutivo (7). Este gen se considera un importante regulador en la morfogénesis ocular que controla los procesos de proliferación, diferenciación y apoptosis celular en el desarrollo normal del ojo. La expresión normal del PAX6 se considera esencial para el desarrollo de tejidos oculares como el epitelio corneal, el iris, el cuerpo ciliar, el cristalino y la retina (8-10).
El papel regulador del gen PAX6 continúa en la vida adulta y se produce a varios niveles.
Regulación de la proliferación y diferenciación del epitelio corneal
La falta de un marcador selectivo de las células madre del limbo (CML), dificulta el estudio del funcionamiento del compartimento proliferativo epitelial en los modelos animales. Los resultados obtenidos con el factor de transcripción nuclear p63, sugerido como posible marcador de células madre epiteliales y de otras más diferenciadas como células transientes amplificadas, no parece que muestren alteraciones en los modelos de mutación PAX6. Tampoco se obtienen resultados concluyentes con marcadores de proliferación celular como el BrdU (11-13). La expresión de CK-12 y CK-3 por las células epiteliales se considera un marcador de diferenciación de estirpe corneal y esta está regulada por el PAX6. Se trata de proteínas del citoesqueleto celular insolubles en agua que forman los filamentos intermedios de las células epiteliales y cuya función fundamental es dar estabilidad a la superficie corneal. En los modelos experimentales de aniridia se observa una expresión disminuida de estas citoqueratinas lo que se traduce en vacuolización y fragilidad del epitelio corneal (14,15).
Regulación de la adhesión celular
Las células del epitelio corneal tienen numerosos mecanismos de adhesión tanto intercelulares como entre éstas y la matriz extracelular. Estos mecanismos incluyen «tight-junctions», «gap-junctions», desmosomas y uniones adherentes. Además, existe una gran variedad de moléculas de adhesión como cateninas, integrinas, desmogleina, desmocolinas que confieren al epitelio corneal una gran resistencia frente a agresiones externas.
En los modelos experimentales de aniridia se observa una reducción en los niveles de desmogleina así como de beta y alfa catenina, proteínas cuya síntesis parece estar regulada por el gen PAX6, lo que conlleva el que aparezcan espacios entre las células epiteliales (16). Estos hallazgos unidos al déficit de citoqueratinas CK-3 y CK-12 conducen a una considerable fragilidad de la superficie corneal que se va a manifestar clínicamente con la presencia de erosiones recurrentes o defectos epiteliales persistentes (17).
Regulación de la actividad de la matriz extracelular
La matriz extracelular proporciona organización estructural a la córnea. En condiciones normales la matriz extracelular está sometida a una lenta pero continua remodelación que se acelera en los procesos de reparación de heridas corneales (18,19). Existe un equilibrio entre la producción de colágeno por los queratocitos y su degradación por un grupo de enzimas denominadas metaloproteinasas (MMP). Estas enzimas son producidas por células epiteliales y fibroblastos y su expresión está influenciada por genes, factores de crecimiento, inhibidores tisulares de metaloproteinasas y constituyentes de las heridas. A su vez, estas enzimas están implicadas en la activación de citoquinas, en la ruptura de moléculas de adhesión y en la creación de fragmentos biológicamente activos (20,21). Se conocen tres tipos de metaloproteinasas: la MMP-1 o colagenasa tipo 1, la MMP-3 o estromelisina y la gelatinasa. Todas ellas se secretan como proenzimas y requieren zinc como cofactor (22). La MMP-1 es producida principalmente por los queratocitos y degrada el colágeno tipo I, II y III. La MMP-3 presenta una intensa actividad proteolítica frente a la caseína, fibronectina y proteoglicanos. La gelatinasa presenta más selectividad por la gelatina y colágeno tipo IV, V y VII. Se distinguen dos tipos de gelatinasas: La MMP-2 es más activa frente al colágeno tipo IV, mientras que la MMP-9 es más activa frente al colágeno tipo V.
La regulación de la Gel-B o MMP-9 depende del PAX6. En los modelos animales de mutación del PAX6, con deficiencia de Gel-B, se observa un acúmulo de fibrina e infiltración por células inflamatorias en relación a un incremento en los niveles de IL-1 (23). El acúmulo de fibrina altera la ordenada disposición de las fibras de colágeno produciendo una pérdida de la transparencia corneal. Por otro lado, la infiltración celular se traduce en un importante estímulo de proliferación neovascular.
En la figura 3 , quedan reflejadas las distintas acciones del gen PAX6 sobre la superficie ocular y su trascendencia clínica.
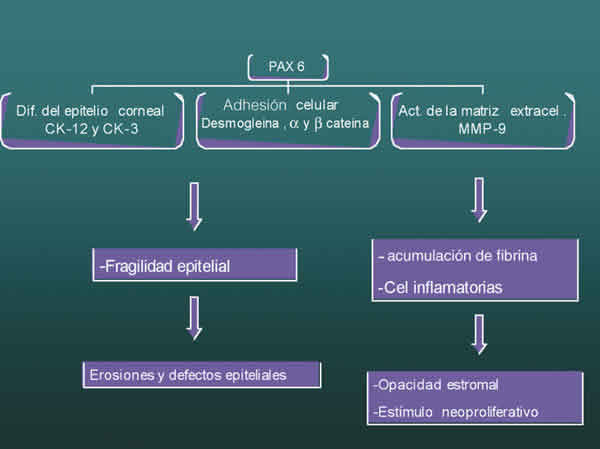
Fig. 3. Patogenia de la queratopatía asociada a la aniridia.
Alteraciones de la superficie ocular en pacientes con aniridia
Cada vez con más frecuencia encontramos en nuestra práctica clínica alteraciones de la superficie ocular motivadas por un síndrome de insuficiencia limbal, que en la mayoría de los casos es producido por agentes externos ya sean traumáticos o inflamatorios. En el caso de la QAA se produce una afectación limbal intrínseca que altera de forma cualitativa la funcionalidad de las CML. Las alteraciones de la superficie ocular que aparecen en la anirida están debidas, al menos inicialmente, a la disfunción de las CML (24). Al igual que en otros casos de insuficiencia limbal parcial, los pacientes con aniridia pueden permanecer asintomáticos hasta que un factor externo actúa sobre el limbo descompensando el lábil equilibrio que mantenía la integridad del epitelio corneal. Así, están descritos agravamientos importantes de los síntomas de QAA tras cirugías con manipulación excesiva del limbo o con aplicación de antimetabólitos tópicos en el control del frecuente glaucoma que asocian (25). Otros autores remarcan la asimetría observada en las alteraciones corneales en estos pacientes y la relaciona, en muchos casos, con antecedentes traumáticos o quirúrgicos en el ojo con peor evolución.
La clínica va a depender, por tanto, del grado de afectación. El espesor de la cornea central suele estar notablemente aumentado (26), siendo común la presencia de neovascularización superficial, fibrosis subepitelial, opacidad corneal progresiva, gran inestabilidad e irregularidad de la superficie ocular que a nivel clínico se traduce en defectos epiteliales persistentes o recurrentes, conjuntivalización corneal, hiperplasia del epitelio limbal, inflamación crónica, hiperemia conjuntival, cambios en la membrana de Bowman y queratinización. Los síntomas incluyen lagrimeo, dolor, disminución de la visión, ojo seco, fotofobia y blefaroespasmo, siendo frecuentes las sobreinfecciones bacterianas y el riesgo de perforación ocular (27,28).
La inestabilidad de la superficie ocular que se produce por la deficiente regeneración del epitelio corneal, va a determinar, entre otras cosas, la aparición de un ojo seco secundario y la pérdida de la función de barrera del epitelio corneal (fig. 4 ). Esta agresión crónica sobre los epitelios de la superficie ocular va a generar una respuesta de éstos en forma de transformación metaplásica. Así pues, la metaplasia escamosa es un proceso utilizado por lo epitelios húmedos para superar temporalmente las agresiones a que se ven sometidos. Este proceso de metaplasia escamosa afecta tanto a las células epiteliales de córnea y conjuntiva, como a las células secretoras (células caliciformes) de la conjuntiva (29). Se trata de una transición patológica y reversible del epitelio normal estratificado no queratinizado a un epitelio queratinizado.

Fig. 4. Tinción con fluoresceína sódica.
Se aprecia la inestabilidad de la película lagrimal y un defecto epitelial.
Clínicamente el proceso de queratinización es fácilmente reconocido mediante biomicroscopía, pero la metaplasia escamosa, previa a la queratinización, puede pasar desapercibida, sobre todo, en los estadios iniciales. En estos casos, la citología de impresión aporta una estimación objetiva del grado de afectación de la superficie ocular, permitiendo un mejor abordaje terapéutico de la enfermedad (30). Además de valorar el grado de metaplasia escamosa, la citología va a permitir identificar la presencia de células caliciformes en el epitelio corneal que es un signo característico de conjuntivalización y por tanto de insuficiencia limbal (fig. 5 ). También mediante citología de impresión se puede estudiar el fenotipo epitelial mediante marcaje con anticuerpos monoclonales de citoqueratinas selectivas de cada linaje celular (31,32). Así, las células del epitelio conjuntival presentan la citoqueratina CK19, mientras que las células epiteliales corneales y limbales presentan las citoqueratinas CK3 y CK12 (33). Las córneas de los pacientes con deficiencia de CML presentan una disminución de las citoqueratinas CK3 y CK12 y un aumento de la citoqueratina CK19 (34). La citología de impresión también permite estudiar la superficie ocular después de intervenciones como trasplante de limbo y trasplante de membrana amniótica (35,36).

Fig. 5. Citología de impresión. La presencia de células caliciformes en la citología de
impresión corneal define la existencia de conjuntivalización del epitelio de la córnea.
Tratar de clasificar todos estos signos y síntomas es importante de cara a planificar el manejo terapéutico de estos pacientes. Nosotros diferenciamos cuatro niveles o estadios de gravedad. Se considera que un paciente presenta clínica de insuficiencia limbal leve (estadio 1) cuando el paciente refiere un máximo de dos episodios de úlceras recurrentes o erosiones recidivantes en los últimos 6 meses, leve fotofobia y epífora y muestran un pannus vascular leve, que no pasa más de 1 mm de la arcada límbica, y mínimos trastornos en la captación de fluoresceína. Se considera que un paciente presenta clínica de insuficiencia limbal moderada (estadio 2) cuando el número de episodios de erosiones recidivantes o úlceras recurrentes es igual o superior a 3 episodios en los últimos 6 meses, presentan una inestabilidad permanente de la película lagrimal y un pannus vascular, acompañado o no de tejido fibroso subepitelial, que afecta a menos de la mitad periférica de la córnea, y la fotofobia, epífora y ojo rojo son la norma. Se considera que un paciente presenta clínica de insuficiencia limbal grave (estadio 3) cuando el paciente presenta una vascularización corneal que afecta al centro de la córnea, así como clínica de erosiones corneales e inestabilidad de la película lagrimal permanentes. La fotofobia, epífora y ojo rojo son la norma, así como la pérdida de visión por afectación del eje visual. En el estadio «0» o insuficiencia limbal subclínica incluimos a pacientes con procesos etiológicos susceptibles de insuficiencia limbal que no manifiestan clínica relacionada con ella (fig. 6 ).
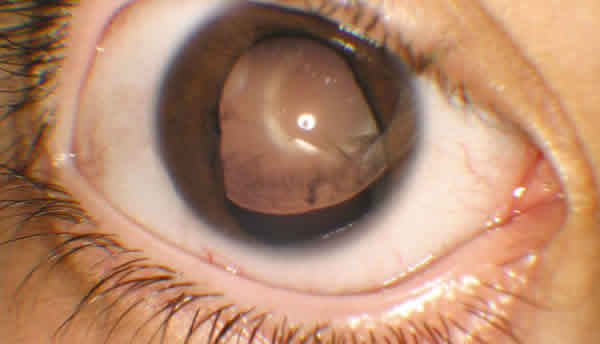
Fig. 6. Queratopatía subclínica en paciente con aniridia.
Tratamiento
Una de las causas de pérdida de visión progresiva y morbilidad en pacientes con aniridia es la queratopatía provocada por la disfunción de las CML. En los últimos años, los avances en el conocimiento de los mecanismos implicados en la renovación celular de la córnea han permitido el correcto abordaje terapéutico de estos pacientes en cualquier fase de la enfermedad (37,38). Aunque, hasta el momento, no ha sido posible demostrar en pacientes con QAA que exista una deficiencia de CML, al no disponer de marcadores específicos que nos permitan identificarlas en los estudios histológicos (39,40), los hallazgos clínicos de estos pacientes coinciden con los descritos en el síndrome de insuficiencia limbar.
Hasta hace unos años, el manejo de la QAA era el tratamiento de soporte con lubricantes tópicos, lentes de contacto terapéuticas o tarsorrafia y cuando desarrollaban opacidad corneal grave se recurría a la queratoplastia lamelar o penetrante con muy malos resultados por recurrencia de las alteraciones corneales prequirúrgicas sobre el injerto (27,41).
Al igual que en el resto de pacientes con insuficiencia limbal, el tratamiento debe ir encaminado a repoblar el limbo esclerocorneal de CML y/o a restaurar el microambiente que las rodea garantizando, así, su expansión y supervivencia.
El manejo terapéutico de estos pacientes dependerá del grado de afectación de la superficie ocular. Así, en pacientes que presenten una insuficiencia limbal subclínica o leve, el tratamiento con lágrimas artificiales sin conservantes puede ser suficiente. Aunque existen muchas lágrimas en el mercado, nosotros preferimos utilizar preparados de hialunorato sódico sin conservantes. Es también importante en estos pacientes una serie de medidas encaminadas a mejorar la sintomatología como la protección solar mediante gafas o favorecer ambientes húmedos. Los episodios de erosiones corneales serán tratados como en cualquier otro paciente mediante oclusión y antibióticos tópicos. En pacientes con queratopatía leve, hemos probado ciclos de tratamiento con suero autólogo encontrando una mejoría subjetiva y una disminución en el número de erosiones corneales (resultado no publicados).
En los pacientes con una queratopatía moderada, el tratamiento con lágrimas artificiales no va a ser suficiente. En estos pacientes se puede recurrir al tratamiento con suero autólogo o al trasplante de membrana amniótica. El tratamiento con suero autólogo se ha mostrado como un arma terapéutica de gran interés en el manejo de distintas patologías que afectan a la superficie ocular (42,43). En el suero autólogo están presentes numerosos factores como el factor de crecimiento epitelial, la fibronectina, la vitamina A, el factor transformador beta de crecimiento de fibroblastos, la a2macroglobulina y factores neurales como la sustancia P, que actúan sobre la proliferación, migración y diferenciación de las células del epitelio corneal (44,45). En este sentido, el suero autólogo ejercería un efecto similar al producido por la membrana amniótica contribuyendo a mejorar el microambiente y facilitando los distintos mecanismos implicados en la renovación y mantenimiento celular de los epitelios de la superficie ocular (46). El trasplante de membrana amniótica (TMA) ofrece grandes ventajas clínicas ya que mejora significativamente el medioambiente en la matriz extracelular de las células epiteliales limbares (47). La utilidad terapéutica de la MA se deriva de sus propiedades mecánicas y, sobre todo, biológicas (48,49). Esto lo hace muy útil en el manejo de pacientes con insuficiencia limbal parcial en donde el aporte de los distintos factores va a favorecer el desarrollo y expansión de las CML supervivientes. El TMA ha sido utilizado en el manejo de la insuficiencia limbal asociado a distintas etiologías (50,51), así como en el tratamiento de la queratopatia asociada a la aniridia (52). Sin embargo, el tratamiento mediante TMA en estos pacientes ofrece sólo resultados transitorios como hemos podido demostrar en pacientes con aniridia (52) (fig. 7 ).

Fig. 7. Trasplante de membrana amniótica. La membrana amniótica cubre
la superficie limbocorneal, suturándose con nylon de 10/0.
En los pacientes con una queratopatía grave o estadio 3, la estabilización de la superficie corneal pasa por aportar CML mediante la realización de un trasplante de limbo (53,54). Debido a que la aniridia se trata de una enfermedad bilateral, la realización de un autotrasplante queda excluida, teniendo que recurrir a alloinjertos con tejido procedente de de familiares sanos, con alta compatibilidad HLA, o de cadáver (55). El riesgo de rechazo, en estos casos, obliga a mantener un tratamiento inmunosupresor oral postoperatorio con potenciales efectos secundarios. El trasplante de limbo procedente de familiares permite una mayor compatibilidad HLA reduciendo, en parte, el riesgo de rechazo, aunque no permite obtener injertos tan extensos como los obtenidos cuando el tejido limbal procede de donante cadáver. La limboqueratoplastia lamelar homóloga se ha realizado en pacientes con aniridia bilateral con aceptables resultados (56). La combinación trasplante de limbo y TMA incrementa el beneficio de cada una de ellas por separado (35,57). Además del trasplante clásico de limbo, en la actualidad podemos aportar CML cultivadas artificialmente sobre membrana amniótica (58-60). La principal ventaja de esta técnica es que se puede transplantar un importante número de células obtenidas a partir de una pequeña muestra de limbo procedente del propio paciente. De esta forma se reduce considerablemente el trauma quirúrgico en el ojo y disminuyen los riesgos de rechazo del allotrasplante de limbo. Aunque se han descrito buenos resultados con estas técnicas en el manejo de distintas patologías de la superficie ocular (61,62), en la práctica existen importantes cuestiones sin resolver en torno a los principios de esta técnica en el caso concreto de los pacientes con aniridia. Por un lado, no existe un método selectivo y específico para identificar las CML y por otro, como se mencionó antes, en la aniridia parece haber más un trastorno del microambiente que rodea a estas CML que una alteración cuantitativa de ellas. Se precisan, por tanto, estudios sobre supervivencia, movilidad y adherencia de las células epiteliales una vez transplantadas en el ojo receptor de pacientes con aniridia.
En la figura 8 , se representa esquemáticamente el manejo de la superficie ocular en pacientes con QAA.

Fig. 8. Esquema terapéutico del manejo de la superficie
ocular en pacientes con aniridia.
Además de la queratopatía, estos pacientes frecuentemente presentan otras alteraciones del segmento anterior como cataratas, glaucoma u opacidad corneal por lo que en ocasiones se precisará de cirugías como trabeculectomías (63,64), trasplantes de córnea (41), o facoemulsificación con o sin implantes de diafragmas intraoculares (65,66). Especial atención merece el tratamiento médico del glaucoma en estos pacientes. Siempre que sea posible se deben utilizar antiglaucomatosos sin conservantes para evitar el daño que, sobre la superficie ocular, producen ciertos conservantes presentes en los preparados farmacológicos de uso habitual en el tratamiento del glaucoma.
La QAA, aunque no aparece en todos los pacientes, es una frecuente causa de morbilidad, por lo que un adecuado enfoque terapéutico puede ser de gran utilidad para mejorar la calidad de vida de estos pacientes. Podemos concluir diciendo que el paciente con aniridia supone un importante reto para el oftalmólogo ya que a los mencionados trastornos del segmento anterior, con frecuencia, asocia trastornos a nivel de la mácula y nervio óptico, nistagmus y ambliopía que reducen considerablemente la capacidad visual de estos ojos (67). Por otro lado, la fotofobia puede llegar a ser muy invalidante teniendo que recurrir en muchos pacientes a la implantación de prótesis de iris (68,69). Por último, no hemos de olvidar que en muchos casos la aniridia se asocia a otros trastornos sistémicos a los que también se debe prestar atención (70,71).
Bibliografía
1. Zolog I, Belengeanu V, Marinca S, Soim A. Familial congenital aniridia. Oftalmologia 1997; 41: 326-328. [ Links ]
2. Ndoye Roth PA, de Medeiros Quenum ME, Wane Khouma AM, Dieng M, Ndiaye PA, Diane MH, et al. Aniridia: five case studies. J Fr Ophthalmol 2005; 28: 845-849. [ Links ]
3. Nishida K, Kinoshita S, Ohashi Y, Kuwayama Y, Yamamoto S. Ocular surface abnormalities in aniridia. Am J Ophthalmol 1995; 120: 368-375. [ Links ]
4. Hanson IM, Fletcher JM, Jordan T, Brown A, Taylor D, Adams RJ, et al. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters´ anomaly. Nat Genet 1994; 6: 168-173. [ Links ]
5. Jordan T, Hanson I, Zaletayev D, Hodgson S, Prosser J, Seawright A, et al. The human PAX6 gene is mutated in two patients with aniridia. Nat Gent 1992; 1: 328-332. [ Links ]
6. Chauhan BK, Yang Y, Cveklova K, Cvekl A. Functional properties of natural human PAX6 and PAX6(5a) mutants. Invest Ophthalmol Vis Sci 2004; 45: 385-392. [ Links ]
7. Callaerts P, Halder G, Gerhring WJ. PAX-6 in development and evolution. Annu Rev Neurosci 1997; 20: 483-532. [ Links ]
8. Collinson JM, Hill RE, West JD. Different roles for Pax6 in the optic vesicle and facial epithelium mediate early morphogenesis of the murine eye. Development 2000; 127: 945-956. [ Links ]
9. Collinson JM, Quinn JC, Buchanan MA, Kaufman MH, Wedden SE, West JD, et al. Primary defects in the lens underlie complex anterior segment abnormalities of the Pax6 heterozygous eye. Proc Natl Acad Sci USA 2001; 98: 9688-9693. [ Links ]
10. Koroma BM, Yang JM, Sundin OH. The Pax-6 homeobox gene is expressed throughout the corneal and conjunctival epithelia. Invest Ophthalmol Vis Sci 1997; 38: 108-120. [ Links ]
11. Collinson JM, Chanas SA, Hill RE, West JD. Corneal development, limbal stem cell function, and corneal epithelial cell migration in the Pax6 (±) mouse. Invest Ophthalmol Vis Sci 2004; 45: 1101-1108. [ Links ]
12. Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A 2001; 98: 3156-3161. [ Links ]
13. Ramaesh K, Dhillon B. Ex vivo expansion of corneal limbal epithelial/stem cells for corneal surface reconstruction. Eur J Ophthalmol 2003; 13: 515-524. [ Links ]
14. Liu CY, Zhu G, Westerhausen-Larson A, Converse R, Kao CW, Sun TT, et al. Cornea- specific expression of K12 keratin during mouse development. Curr Eye Res 1993; 12: 963-974. [ Links ]
15. Chiambaretta F, Blanchon L, Rabier B, Kao WW, Liu JJ, Dastugue B, et al. Regulation of corneal keratin-12 gene expression by the human Kruppel-like transcription factor 6. Invest Ophthalmol Vis Sci 2002; 43: 3422-3429. [ Links ]
16. Davis J, Duncan MK, Robison WG Jr, Piatigorsky J. Requirement for Pax6 in corneal morphogenesis: a role in adhesion. J Cell Sci 2003; 116: 2157-2167. [ Links ]
17. Ramaesh T, Collinson JM, Ramaesh K, Kaufman MH, West JD, Dhillon B. Corneal abnormalities in Pax6+/- small eye mice mimic human aniridia-related keratopathy. Invest Ophthalmol Vis Sci 2003; 44: 1871-1878. [ Links ]
18. Fini ME. Keratocyte and fibroblast phenotypes in the repairing cornea. Prog Retin Eye Res 1999; 18: 529-551. [ Links ]
19. Jester JV, Petroll WM, Barry PA, Cavanagh HD. Expression of alpha-smooth muscle (alpha-SM) actin during corneal stromal wound healing. Invest Ophthalmol Vis Sci 1995; 36: 809-819. [ Links ]
20. Sivak JM, Mohan R, Rinehart WB, Xu PX, Maas RL, Fini ME. Pax-6 expression and activity are induced in the reepithelializing cornea and control activity of the transcriptional promoter for matrix metalloproteinase gelatinase B. Dev Biol 2000; 222: 41-54. [ Links ]
21. Sivak JM, Fini ME. MMPs in the eye: emerging roles for matrix metalloproteinases in ocular physiology. Prog Retin Eye Res 2002; 21: 1-14. [ Links ]
22. Brown D, Chwa M, Escobar M, Kenney MC. Characterization of the major matrix degrading metalloproteinase of human corneal stroma. Evidence for an enzyme/inhibitor complex. Exp Eye Res 1991; 52: 5-16. [ Links ]
23. Mohan R, Chintala SK, Jung JC, Villar WV, McCabe F, Russo LA, et al. Matrix metalloproteinase gelatinase B (MMP-9) coordinates and effects epithelial regeneration. J Biol Chem 2002; 277: 2065-2072. [ Links ]
24. Dua HS, Saini JS, Azuara-Blanco A, Gupta P. Limbal stem cell deficiency: concept, aetiology, clinical presentation, diagnosis and management. Indian J Ophthalmol 2000; 48: 83-92. [ Links ]
25. Nelson LB, Spaeth GL, Nowinski TS, Margo CE, Jackson L. Aniridia. A review. Surv Ophthalmol 1984; 28: 621–642. [ Links ]
26. Brandt JD, Casuso LA, Budenz DL. Markedly increased central corneal thickness: an unrecognized finding in congenital aniridia. Am J Ophthalmol 2004; 137: 348-350. [ Links ]
27. Gomes JA, Eagle RC Jr, Gomes AK, Rapuanao CJ, Cohen EJ, Laibson PR. Recurrent keratopathy after penetrating keratoplasty for aniridia. Cornea 1996; 15: 457-462. [ Links ]
28. Jastanciah S, Al-Rajhi AA. Association of aniridia and dry eyes. Ophthalmology 2005; 112: 1535-1540. [ Links ]
29. Rivas L, Murube J, Rivas A, Murube E. Estudio del ojo seco en pacientes con aniridia congénita, mediante citología de ipresión. Arch Soc Esp Oftalmol 2003; 78: 615-622. [ Links ]
30. Sridhar MS, Vemuganti GK, Bansal AK, Rao GN. Impression cytology-proven corneal stem cell deficiency in patients after surgeries involving the limbus. Cornea 2001; 20: 145-148. [ Links ]
31. Lavker RM, Tseng SC, Sun TT. Corneal epithelial stem cells at the limbus: looking at some old problems from a new angle. Exp Eye Res 2004; 78: 433-446. [ Links ]
32. Buck RC. Ultrastructure of conjunctival epithelium replacing corneal epithelium. Curr Eye Res 1986; 5: 149-159. [ Links ]
33. Lauweryns B, van den Oord JJ, Missotten L. The transitional zone between limbus and peripheral cornea. An immunohistochemical study. Invest Ophthalmol Vis Sci 1993; 34: 1991-1999. [ Links ]
34. Tseng SC, Li DQ. Comparison of protein kinase C subtype expression between normal and aniridic human ocular surfaces: implications for limbal stem cell dysfunction in aniridia. Cornea 1996; 15: 168-178. [ Links ]
35. Lopez-Garcia JS, Rivas L, Garcia-Lozano I. Tratamiento de la insuficiencia limbal grave mediante cirugía combinada de trasplante de limbo y trasplante de membrana amniótica. Arch Soc Esp Oftalmol 2005; 80: 405-412. [ Links ]
36. Prabhasawat P, Tseng SC. Impression cytology study of epithelial phenotype of ocular surface reconstructed by preserved human amniotic membrane. Arch Ophthalmol 1997; 115: 1360-1367. [ Links ]
37. Dua HS, Azuara-Blanco A. Limbal stem cells of the corneal epithelium. Surv Ophthalmol 2000; 44: 415-425. [ Links ]
38. Tseng SC. Concept and application of limbal stem cells. Eye 1989; 3: 141-157. [ Links ]
39. Schlotzer-Schrehardt U, Kruse FE. Identification and characterization of limbal stem cells. Exp Eye Res 2005; 81: 247-264. [ Links ]
40. Ramaesh K, Ramaesh T, Dutton GN, Dhillon B. Evolving concepts on the pathogenic mechanisms of aniridia related keratopathy. Int J Biochem Cell Biol 2005; 37: 547-557. [ Links ]
41. Tiller AM, Odenthal MT, Verbraak FD, Gortzak-Moorstein N. The influence of keratoplasty on visual prognosis in aniridia: a historical review of one large family. Cornea 2003; 22: 105-110. [ Links ]
42. Geerling G, Maclennan S, Hartwig D. Autologuos serum eye drops for ocular surface disorders. Br J Ophthalmol 2004; 88: 1467-1474. [ Links ]
43. Kojima T, Ishida R, Dogru M, Goto E, Matsumoto Y, Kaido M, et al. The effect of autologous serum eyedrops in the treatment of severe dry eye disease: a prospective randomized case-control study. Am J Ophthalmol 2005; 139: 242-246. [ Links ]
44. Poon AC, Gerling G, Dart JK, Fraenkel GE, Daniels JT. Autologuos serum eyedrops for dry eyes and epithelial defects: clinical and in vitro toxicity studies. Br J Ophthalmol 2001; 85: 1188-1197. [ Links ]
45. Alvarado Valero MC, Martinez Toldos JJ, Borras Blasco J, Alminana Alminana A, Pérez Ramos JM. Tratamiento de los defectos epiteliales persistentes mediante suero autólogo. Arch Soc Esp Oftalmol 2004; 79: 537-542. [ Links ]
46. Noble BA, Loh RS, MacLennan S, Pesudovs K, Reynolds A, Bridges LR, et al. Comparison of autologuos serum eye drops with conventional therapy in a randomised controlled crossover trial for ocular disease. Br J Ophthalmol 2004; 88: 647-652. [ Links ]
47. Kim JS, Kim JC, Na BK, Jeong JM, Song CY. Amniotic membrane patching promotes healing and inhibits proteinase activity on wound healing following acute corneal alkali burn. Exp Eye Res 2000; 70: 329-337. [ Links ]
48. Koizumi NJ, Inatomi TJ, Sotozono CJ, Fullwood NJ, Quantock AJ, Kinoshita S. Growth factor mRNA and protein in preserved human amniotic membrane. Curr Eye Res 2000; 20: 173-177. [ Links ]
49. Sonnenberg A, Calafat J, Janssen H, Daams H, van der Raaij-Helmer LM, Falcioni R, et al. Integrin alpha 6/beta 4 complex is located in hemidesmosomes, suggestin a major role in epidermal cell-basement membrane adhesion. J Cell Biol 1991; 113: 907-917. [ Links ]
50. Sridhar MS, Bansal AK, Sangwan VS, Rao GN. Amniotic membrane transplantation in acute chemical and thermal injury. Am J Ophthalmol 2000; 130: 134-137. [ Links ]
51. Pires RT, Tseng SC, Prabhasawat P, Puangsricharern V, Maskin SL, Kim JC, et al. Amniotic membrane transplantation for symptomatic bullous keratopathy. Arch Ophthalmol 1999; 117: 1291-1297. [ Links ]
52. López-García JS, Rivas L, García-Lozano I. Trasplante de membrana amniótica en el tratamiento de la insuficiencia limbal moderada de pacientes con aniridia congénita. Arch Soc Esp Oftamol 2005; 80: 517-523. [ Links ]
53. Kruse FE, Reinhard T. Limbus transplantation for reconstruction of the ocular surface. Ophthalmologe 2001; 98: 818-831. [ Links ]
54. Holland EJ, Djalilian AR, Schwartz GS. Management of aniridic keratopathy with keratolimbal allograft: a limbal stem cell transplantation technique. Ophthalmology 2003; 110: 125-130. [ Links ]
55. Meisler DM, Perez VL, Proudfit J. A device to facilitate limbal stem cell procurement from eye bank donor tissue for keratolimbal allograft procedures. Am J Ophthalmol 2005; 139: 212-214. [ Links ]
56. Sundmacher R, Reinhard T. Homologous lamellar central limbokeratoplasty in severe limbal stem cell deficiency. Klin Monatsbl Augenheilkd 1998; 213: 254-255. [ Links ]
57. Tseng SC, Prabhasawat P, Barton K, Gray T, Meller D. Amniotic membrane transplantation with or without allografts for corneal surface reconstruction in patients with limbal stem cells deficiency. Arch Ophthalmol 1998; 116: 431-441. [ Links ]
58. Koizumi N, Inatomi T, Quantock AJ, Fullwood NJ, Dota A, Kinoshita S. Amniotic membrane as a substrate for cultivating limbal corneal epithelial cells for autologous transplantation in rabbits. Cornea 2000; 19: 65-71. [ Links ]
59. Schwab IR, Reyes M, Isseroff RR. Successful transplantation of bioengineered tissue replacements in patients with ocular surface disease. Cornea 2000; 19: 421-426. [ Links ]
60. Tsai RJ, Li LM, Chen J. Reconstruction of damaged corneas by transplantation of autologous limbal epithelial cells. N Engl J Med 2000; 343: 86-93. [ Links ]
61. Shimazaki J, Shinozaki N, Tsubota K. Transplantation of amniotic membrane and limbal autograft for patients with recurrent pterygium associated with symblepharon. Br J Ophthalmol 1998; 82: 235-240. [ Links ]
62. Nakamura T, Kinoshita S. Ocular surface reconstruction using cultivated mucosal epithelial stem cells. Cornea 2003; 22: S75-S80. [ Links ]
63. Arroyave CP, Scott IU, Gedde SJ, Parrish RK 2nd, Feuer WJ. Use of glaucoma drainage devices in the management of glaucoma associated with aniridia. Am J Ophthalmol 2003; 135: 155-159. [ Links ]
64. Chen TC, Walton DS. Goniosurgery for prevention of aniridic glaucoma. Arch Ophthalmol 1999; 117: 1144-1148. [ Links ]
65. Mavrikakis I, Casey JM. Phacoemulsification and endocapsular implantation of an artificial iris intraocular lens in traumatic cataract and aniridia. J Cataract Refrac Surg 2002; 28: 1088-1091. [ Links ]
66. Esquenazi S, Amador S. Bilateral cataract surgery combined with implantation of a brown diaphragm intraocular lens after trabeculectomy for congenital aniridia. Ophthalmic Surg Lasers 2002; 33: 514-517. [ Links ]
67. McCulley TJ, Mayer K, Dahr SS, Simpson J, Holland EJ. Aniridia and optic nerve hypoplasia. Eye 2005; 19: 762-764. [ Links ]
68. Menezo JL, Martinez-Costa R, Cisneros A, Desco MC. Implantation of iris devices in congenital and traumatic aniridias: surgery solutions and complications. Eur J Ophthalmol 2005; 15: 451-457. [ Links ]
69. Mavrikakis I, Mavrikakis E, Syam PP, Bell J, Casey JH, Casswell AG, et al. Surgical management of iris defects with prosthetic iris devices. Eye 2005; 19: 205-209. [ Links ]
70. Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M. WAGR syndrome: a clinical review of 54 cases. Pediatrics 2005; 116: 984-988. [ Links ]
71. Yasuda T, Kajimoto Y, Fujitani Y, Watada H, Yamamoto S, Watarai T, et al. PAX6 mutations as a genetic factor common to aniridia and glucose intolerance. Diabetes 2002; 51: 224-230. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
J.S. López García
Servicio de oftalmología
Hospital Central de Cruz Roja
Avenida Reina Victoria, 26
28003 Madrid, España.
e-mail: docsantilopez@hotmail.com
Recibido: 07.02.06
Aceptado: 24.07.06

