










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versão impressa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.83 no.6 Jun. 2008
Síndrome de Vogt Koyanagi Harada
Vogt Koyanagi Harada Syndrome
De-Domingo B.1, Blanco M.J.2, Rodríguez-Cid M.J.2, Piñeiro A.2, Mera P.1, Capeáns M.C.2
Complejo Hospitalario Universitario de Santiago de Compostela. Servicio de Oftalmología. Santiago de Compostela. España.
1 Licenciado en Medicina.
2 Doctor en Medicina.
Dirección para correspondencia
RESUMEN
Casos clínicos: Caso 1. Mujer con pérdida visual y cefalea. Presentaba un desprendimiento de retina exudativo bilateral. Se inició tratamiento corticoideo y posteriormente se añadió ciclosporina-A. Tras nueve meses, su visión es de 10/10 en ojo derecho y 8/10 en izquierdo.
Caso 2. Mujer con visión borrosa. Mostraba vitritis con desprendimiento seroso bilateral. Tras iniciar tratamiento corticoideo permanece estable con visión de 9/10 en ojo derecho y 8/10 en izquierdo.
Discusión: El Síndrome de Vogt Koyanagi Harada es una panuveítis bilateral acompañada de alteraciones neurológicas y cutáneas.
La sospecha ante desprendimientos de retina exudativos, multifocales y bilaterales con sintomatología sistémica es fundamental para el diagnóstico.
Su tratamiento precoz y mantenido es la base de una buena evolución.
Palabras clave: Panuveítis bilateral, síndrome de Harada, desprendimiento seroso, uveomeningoencefalitis, síndrome de Vogt-Koyanagi.
ABSTRACT
Case reports: Case 1. A female patient with visual loss and migraine who was found to have an exudative, bilateral retinal detachment. Steroid therapy was commenced initially and then cyclosporin-A added. Nine months later, her vision was 10/10 in the right eye and 9/10 in the left eye. Case 2. A female patient with unclear vision who was found to have an exudative, bilateral retinal detachment. Systemic steroid therapy was given and ultimately her vision was 9/10 in the right eye and 8/10 in the left eye.
Discussion: Vogt Koyanagi Harada syndrome is a bilateral granulomatous panuveitis associated with neurologic and dermatologic disorders. When a patient has exudative multifocal and bilateral retinal detachment and systemic symptomatology, the possibility of this disease needs to be suspected. Good visual prognosis is possible if early treatment is given (Arch Soc Esp Oftalmol 2008; 83: 385-390).
Key words: Bilateral panuveitis, Harada syndrome, serous detachment, uveomeningoen, Vogt Koyanagi Harada syndrome.
Introducción
El síndrome de Vogt-Koyanagi-Harada (VKH) es un transtorno multisistémico e idiopático que afecta de forma característica a individuos pigmentados, preferentemente a mujeres jóvenes (1). Se trata de una panuveítis granulomatosa bilateral difusa que se acompaña de alteraciones neurológicas, auditivas y cutáneas. Se ha descrito que la positividad en sangre del haplotipo HLA-DRB1*0405 aumenta la susceptibilidad de padecer la enfermedad. Su patogenia se ha relacionado con alteraciones inmunológicas cuya célula diana es el melanocito.
A continuación se exponen dos casos de VKH diagnosticados y tratados en nuestro centro.
Caso clínico
Caso clínico 1
Mujer de 28 años, con antecedentes de «Síndrome de múltiples puntos blancos evanescentes» en ambos ojos (fig. 1) hace 7 años y buena respuesta a corticoterapia oral. Acude a urgencias de nuestro hospital por disminución de agudeza visual bilateral y cefalea de 15 días de evolución. A la exploración oftalmológica presenta una agudeza visual de 3/10 con reacción inflamatoria en cámara anterior y desprendimiento de retina exudativo bilateral (fig. 2). Se inicia tratamiento corticoideo oral a altas dosis (80 mg de prednisona), constatándose una buena respuesta inicial. Al mes del tratamiento se observa una recidiva del desprendimiento de retina foveal en el ojo izquierdo, por lo que se asocia a su tratamiento CyA (dosis inicial de 4 mg/kg/día). Tras nueve meses de seguimiento, presenta una agudeza visual de unidad en el ojo derecho y de 8/10 en el ojo izquierdo con movilización pigmentaria en cuatro cuadrantes y fibrosis subretiniana (fig. 3).
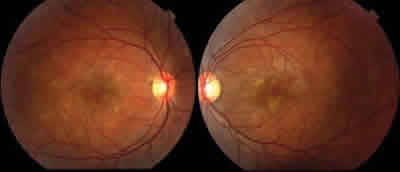
Fig. 1: Síndrome de múltiples puntos blancos evanescentes bilateral.
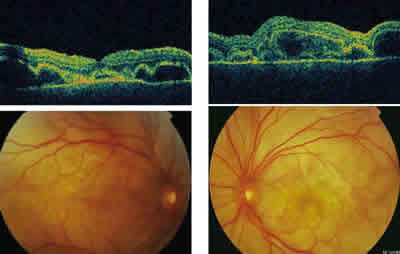
Fig. 2: Imagen biomicroscópica y tomografía óptica de coherencia
del desprendimiento de retina exudativo bilateral.
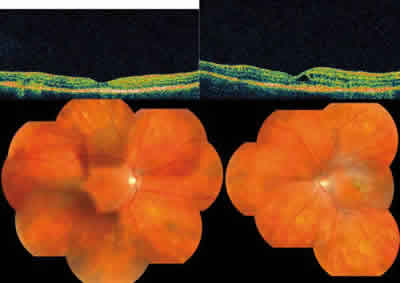
Fig. 3: Movilización pigmentaria en cuatro cuadrantes y fibrosis
subretiniana en ojo izquierdo.
Caso clínico 2
Mujer de 45 años, sin antecedentes oftalmológicos de interés. Acude a urgencias por náuseas, visión borrosa y dolor en ambos ojos. A la exploración oftalmológica presenta una agudeza visual de movimiento de manos en ambos ojos con inflamación en cámara anterior y vitritis con desprendimiento seroso que afecta a polo posterior de ambos ojos (fig. 4).

Fig. 4: Imagen biomicroscópica de desprendimientos
de retina exudativos bilaterales.
Es enviada a nuestro servicio para valoración y tratamiento. Tras iniciar tratamiento corticoideo a altas dosis (200 mg intravenosos de prednisolona que al tercer día se reducen paulatinamente a 160 mg, 120 mg, 80 mg y 60 mg cada tres días, pasando posteriormente a tratamiento con prednisona vía oral), se realiza una angiografía fluoresceínica y un OCT de control. La paciente permanece estable durante el primer año de seguimiento con tratamiento, presentando una agudeza visual de 9/10 en ojo derecho y 8/10 en ojo izquierdo (fig. 5). En el fondo de ojo se observa una atrofia generalizada del epitelio pigmentario con movilización de pigmento (fig. 6).
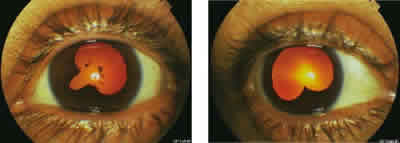
Fig. 5: Imagen de segmento anterior de la paciente tras
el primer año de seguimiento.
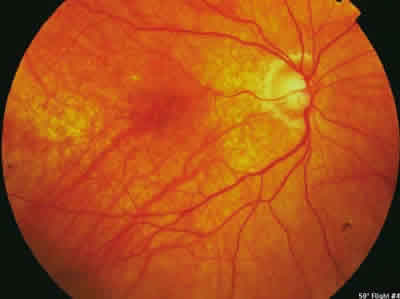
Fig. 6: Movilización pigmentaria generalizada y acúmulo de pigmento.
Discusión
El diagnóstico del VKH es fundamentalmente clínico, siendo necesario el diagnóstico diferencial con la oftalmía simpática, el linfoma primario de células B, la escleritis posterior y el síndrome de efusión uveal. La evolución de la enfermedad podemos dividirla en cuatro estadios clínicos: prodrómico, oftalmológico, de convalecencia y crónico recurrente.
En el estadio prodrómico, los pacientes muestran cefalea, naúseas, vértigo... encontrándose pleocitosis en el líquido cefalorraquídeo. En unos días comienza el estadio oftalmológico con visión borrosa o escotoma central, siendo inicialmente bilateral en el 80% de los casos. El hallazgo ocular más característico es el desprendimiento de retina exudativo multifocal. En la angiografía fluoresceínica (AGF) se observa el escape de colorante desde la coroides al espacio subretiniano en múltiples zonas, sin escape de colorante en los vasos retinianos. La neovascularización en estos pacientes se produce tanto por la inflamación mantenida, como por la isquemia coroidea, siendo fundamental la AGF para diferenciar ambos procesos y tratarlos de manera adecuada.
En el estadio de convalecencia se produce la despigmentación de los tejidos, apareciendo en la fase crónica recurrente las uveítis recidivantes y las complicaciones oftalmológicas.
Los criterios diagnósticos del VKH son válidos para el diagnóstico de certeza en los estadíos finales de la enfermedad, no así en las fases iniciales, por lo que sería necesario llevar acabo una revisión de los mismos (2).
Se ha descrito el KU=MEL=1 en el HLA=DRB1 como un nuevo autoantígeno para el VKH, sugiriéndose su posible uso en el diagnóstico y tratamiento de esta enfermedad (3).
El tratamiento fundamental son los corticoides sistémicos y tópicos. La dosis inicial, la duración del tratamiento y la velocidad de retirada, deben individualizarse para cada paciente (4,5).
El pronóstico visual de los pacientes es generalmente bueno si el diagnóstico es temprano y se prescribe un tratamiento adecuado de forma agresiva y mantenida en el tiempo.
Bibliografía
1. Yang P, Ren Y, Li B, Fang W, Meng Q, Kijlstra A. Clinical characteristics of Vogt-Koyanagi-Harada syndrome in Chinese patients. Ophthalmology 2007; 114: 606-614. [ Links ]
2. Tsai JH, Evans M, Rao NA. Comparative study of two sets of criteria for the diagnosis of Vogt-Koyanagi-Harada disease. Am J Ophthalmol 2006; 141: 778-779. [ Links ]
3. Otani S, Sakurai T, Yamamoto K, Fujita T, Matsuzaki Y, Goto Y, et al. Frequent immune response to a melanocyte specific protein KU-MEL-1 in patients with Vogt-Koyanagi-Harada disease. Br J Ophthalmol 2006; 90: 773-777. [ Links ]
4. Touitou V, Escande C, Bodaghi B, Cassoux N, Wechsler B, Lemaitre C, et al. Diagnostic and therapeutic management of Vogt-Koyanagi-Harada syndrome. J Fr Ophtalmol 2005; 28: 9-16. [ Links ]
5. Read RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, et al. Evaluation of the effect on outcomes of the route of administration of corticosteroids in acute Vogt-Koyanagi-Harada disease. Am J Ophthalmol 2006; 142: 119-124. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Begoña de Domingo Barón
Moas de Abaixo, 4 C
15897 Santiago de Compostela (La Coruña)
España
E-mail: bdedbar@yahoo.es
Recibido: 18/5/07.
Aceptado: 30/4/08.

