










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.83 no.12 dic. 2008
Vitreorretinopatía exudativa familiar: nuestra experiencia
Familial exudative vitreoretinopathy: our experience
Piñero A.M.1, Sempere J.1, Nadal J.1, Elizalde-Montagut J.2
Instituto Universitario Barraquer. Barcelona. España.
1 Licenciado en Medicina.
2 Doctor en Medicina.
Comunicación presentada parcialmente en el LXXXII Congreso de la S.E.O. (A Coruña 2006).
Dirección para correspondencia
RESUMEN
Objetivo: Evidenciar la variabilidad de la presentación inicial de la vitreorretinopatía exudativa familiar (VREF) y la importancia de su diagnóstico y tratamiento precoz. Así como, valorar la respuesta al tratamiento de los diferentes estadios clínicos.
Métodos: Realizamos un estudio retrospectivo, intervencional y descriptivo de 11 pacientes (17 ojos) afectos de vitreorretinopatía exudativa familiar tratados en nuestro centro.
Resultados: De los pacientes afectos de vitreorretinopatía exudativa familiar, un 33,3% se encontraban en el estadio I, un 16,6% en el estadio II y el 50% restante en estadio III.
Los resultados tras el tratamiento fueron los siguientes. En los estadios I y II, la agudeza visual se mantuvo estable (agudeza visual media 0,8 y 0,4 respectivamente). En el estadio III se observó un empeoramiento de la agudeza visual.
Conclusiones: El tratamiento en estadios iniciales puede mejorar el pronóstico visual y evitar complicaciones como la exudación subfoveal crónica y la contracción y fibrosis en la interfase retino vítrea periférica que pueden provocar ectopia macular.
Palabras clave: Vitreorretinopatía, retina, exudación, distrofia, vitrectomía.
ABSTRACT
Purpose: To describe our experience in the diagnosis of Familial Exudative Vitreoretinopathy (FEVR) at its different clinical stages. We also report our outcomes in early treatment of this inherited disorder.
Methods: Retrospective interventional and descriptive case series of 11 patients (17 eyes) affected by FEVR evaluated and treated in our hospital.
Results: In our series, 33.3% of patients were classified as stage I, while 16.7% and 50% were classified as stage II and III, respectively.
Visual acuity (VA) remained stable after treatment in stages I and II (mean VA was 0.8 and 0.4, respectively) but decreased in stage III.
Conclusions: Early treatment of FERV may improve the visual prognosis and avoid complications such as subfoveal chronic exudation and vitreoretinal peripheral interphase contraction and fibrosis (Arch Soc Esp Oftalmol 2008; 83: 703-708).
Key words: Vitreoretinopathy, retina, exudation, dystrophy, vitrectomy.
Introducción
La vitreorretinopatía exudativa familiar (VREF) es una enfermedad perteneciente al grupo de las distrofias vitreorretinianas (fig. 1) de baja incidencia y herencia autosómica dominante (1,2). Es más frecuente en la primera década y se presenta de manera bilateral aunque su desarrollo suele ser asimétrico (1).
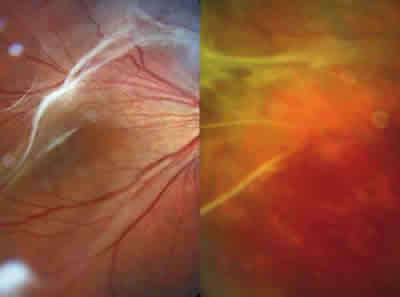
Fig. 1: Distrofia vitreorretiniana caracterizada por sinéresis
y licuefacción vítrea y por la formación de bandas fibrilares.
Se caracteriza fundamentalmente por isquemia periférica (2) y alteraciones en la interfase vitreorretiniana, ambas más frecuentes en la zona temporal (1,2).
Los pacientes pierden visión conforme va avanzando la enfermedad, pudiendo conducir a la ceguera por las complicaciones derivadas de la isquemia y proliferación fibrovascular. Por ello el objetivo primordial es el diagnóstico y tratamiento precoz de la enfermedad.
Se diferencian tres estadios clínicos (3):
– Estadio I: se define como grado leve de la enfermedad en el que los pacientes suelen estar asintomáticos y en el que se observa alteración en la interfase vitreorretiniana, como blanco con o sin presión o degeneración cistoide, y zonas avasculares en la retina periférica. Menos característicos son la ingurgitación vascular, las telangiectasias, los microaneurismas y los shunts arteriovenosos (figs. 2-4).
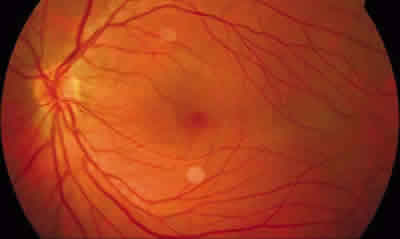
Fig. 2: Retinografía de OI de un paciente con VREF estadio I.
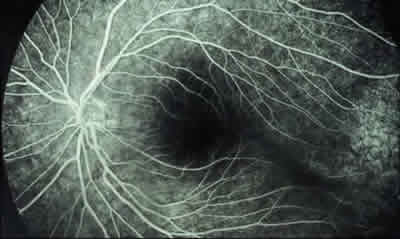
Fig. 3: AGF del caso anterior. Se observa alteración
del EPR temporal a la mácula.
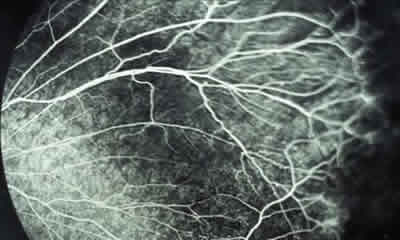
Fig. 4: AGF del mismo caso, en la que se confirma
la isquemia periférica de cuadrantes temporales.
– Estadio II representa el estadio proliferativo y exudativo de la enfermedad. Se puede observar neovascularización, proliferación fibrovascular y exudación sub e intrarretiniana. Las lesiones fibrovasculares pueden provocar complicaciones como ectopia macular y fenómenos de tracción papilar (figs. 5-7).
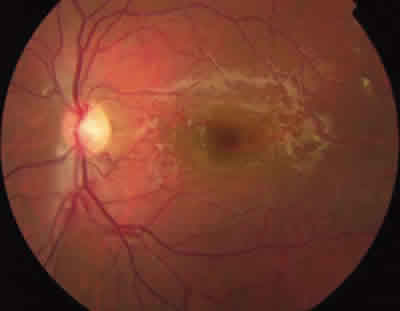
Fig. 5: Paciente en estadio II. Se aprecian pequeñas
proliferaciones fibrovasculares en polo posterior.
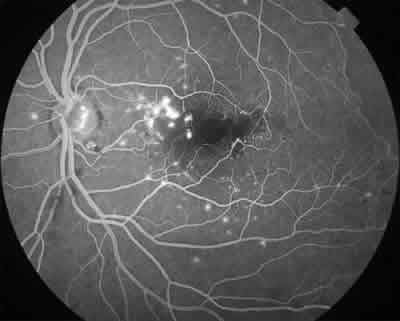
Fig. 6: AGF de polo posterior del mismo caso. Se observa escape
de fluoresceína por alteraciones vasculares telangiectásicas e
isquemia foveal parcial (ruptura de la red capilar perifoveal).
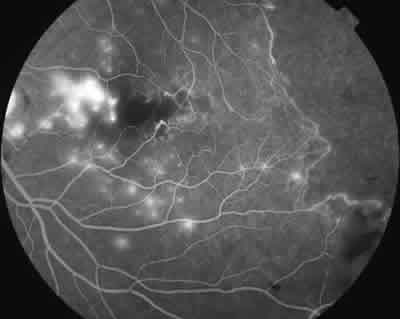
Fig. 7: Mismo caso en el que se ve ruptura de
la red vascular periférica temporal.
– Estadio III la lesión cicatricial causa desprendimiento de retina traccional, regmatógeno y exudativo, y pliegues falciformes.
El cuadro clínico, en ocasiones, se complica con atrofia óptica, catarata, glaucoma y queratopatía en banda (figs. 8-10).

Fig. 8: Ojo en estadio III. Se aprecia tortuosidad vascular,
exudación subretiniana y bandas retinianas traccionales.
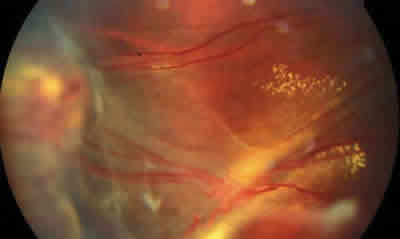
Fig. 9: Otro caso diagnosticado de estadio III en el que se
observa tortuosidad vascular, exudación subretiniana y DR exudativo.
Fig. 10: Catarata polar anterior y banda fibrosa adherida a
cristaloides posterior en paciente diagnosticado de VREF estadio III.
El tratamiento de esta enfermedad varia según el estadio. Así, en el estadio I, en el que los pacientes suelen presentarse asintomáticos, el tratamiento va dirigido a realizar profilaxis mediante fotocoagulación (FCG) y criocoagulación (CCG) (4). En el estadio II el tratamiento debe ir dirigido hacia la liberación de las tracciones vitreorretinianas periféricas mediante la disección de membranas, la colocación de cerclaje escleral y vitrectomía vía pars plana (VPP).
En el estadio III las opciones quirúrgicas se combinan debido al estadio avanzado de la enfermedad (5).
Sujetos, material y métodos
Se presenta un estudio retrospectivo, intervencional y descriptivo de los pacientes afectos de vitreorretinopatía exudativa familiar tratados en nuestro centro.
El objetivo de este estudio es evidenciar la variabilidad de la presentación inicial de la vitreorretinopatía exudativa familiar y la importancia de su diagnóstico y tratamiento precoz. Así como, valorar la respuesta al tratamiento de los diferentes estadios clínicos.
Se estudiaron diecisiete ojos correspondientes a once pacientes.
Resultados
Los pacientes presentaban edades comprendidas entre 6 y 24 años, con una proporción hombre/mujer 3:1.
La distribución de los pacientes en base al estadio fue la siguiente:
El 33,3% de los pacientes se encontraban en el estadio I, el 16,6% de los pacientes se diagnosticaron como estadio II y el 50% restante se encontraban en el estadio III.
El tratamiento que se aplicó a nuestros casos fue el siguiente.
Un 15,3% de los pacientes del estadio I, recibieron una sesión de FCG. El 18% restante recibieron una sesión de FCG y otra de CCG en el área temporal, que se tuvieron que completar a 360° posteriormente. La agudeza visual se mantuvo estable, con una AV media de 0,8.
En el estadio II, todos recibieron una sesión de FCG y con el tiempo acabaron sometiéndose tanto a vitrectomía (VPP) como a cerclaje escleral. También se mantuvo en estos casos la AV estable, con un valor medio de 0,4.
Los casos diagnosticados en estadio III (50%) recibieron varias sesiones de FCG y CCG. También se realizaron VPP, disección de hialoides posterior y membranas y colocación de cerclaje escleral. La AV final en estos casos empeoró (AV media de 0,05).
Discusión
La VREF fue descrita en 1969 por Criswick y Schepens (6). Presentaron seis pacientes con una clínica similar a la que presentan los pacientes con retinopatía del prematuro (ROP) pero sin antecedentes personales de prematuridad ni suplementación de oxígeno.
Describimos un estudio retrospectivo, intervencional y descriptivo de 17 ojos correspondientes a 11 pacientes afectos de VREF. Esta patología al igual que la ROP se caracteriza por la presencia de áreas de isquemia en la periferia de la retina, siendo condición indispensable para el diagnóstico.
En nuestro caso realizamos el diagnóstico de sospecha en base a los hallazgos clínicos; la angiografía fluoresceínica realizada en todos los casos lo confirmó. El 33,3% de nuestros ojos sólo presentaban isquemia retiniana periférica y fueron clasificados en estadio I. En los pacientes que se clasificaron como como estadio II (16,6%) se observaron además proliferación fibrovascular, neovascularización y exudación subretiniana. En el restante 50% de los ojos se apreció un componente cicatricial asociado a un desprendimiento de retina traccional, regmatógeno y exudativo. Por lo tanto es importante conocer la variabilidad de la presentación clínica y realizar el diagnóstico diferencial con otras patologías como la enfermedad de Eales, la retinopatía de la prematuridad y otras que cursen con isquemia periférica.
La VREF se trata de una enfermedad genéticamente heterogénea ya que puede presentarse con diferentes patrones hereditarios (1). El patrón más frecuentemente descrito es el autosómico dominante (AD). También se encuentran en la literatura casos de herencia autosómica recesiva (AR), herencia recesiva ligada al cromosoma X, así como algunos casos esporádicos.
Hay descritas en la literatura diferentes mutaciones genéticas responsables de las distintas formas de herencia de la VREF.
La alteración del gen FZD4 que codifica el receptor Wnt Frizzled-4 y que ocupa el locus EVR1, esta localizado en el cromosoma 11 locus 11q 13-23 y es el responsable de las formas de herencia AD.
El segundo locus o EVR2 corresponde a la alteración del mismo gen responsable de la enfermedad de Norrie. Se ha descrito su mutación en los casos de VREF ligada al cromosoma X.
Otra alteración genética, locus EVR3, es la del gen localizado en el cromosoma 11 locus 11p 12-13 del que todavía se desconocen sus características.
Un cuarto gen presente en el locus EVR4 es el gen LRP5 que codifica un receptor lipoprotéico de baja densidad y que se encuentra localizado en el cromosoma 11 locus 11q13. Éste también es considerado responsable de alguna forma AD.
Por último, las formas AR también han sido descritas aunque todavía se desconoce el defecto genético responsable de las mismas.
El manejo de estos pacientes es controvertido. No todos los autores defienden la profilaxis con fotocoagulación (FCG) o criocoagulación (CCG), a pesar de evidenciar angiográficamente áreas isquémicas en la retina periférica. La indicación principal para el tratamiento profiláctico, según la mayoría de investigadores, es la neovascularización periférica y el desprendimiento de retina exudativo, aunque los resultados no han sido concluyentes (7,8). Un 15,3% de los ojos diagnosticados en el estadio I en nuestra serie, recibieron una sesión de FCG, mientras que el resto recibieron, además, otra sesión de CCG. En los estadios II y III, según del grado de evolución de a enfermedad, las opciones terapéuticas abarcaron desde FCG y CCG, hasta VPP y cerclaje escleral, así como peeling de membranas y disección de las proliferaciones fibrovasculares.
Por lo tanto, creemos que es importante el diagnóstico en los estadios precoces para mejorar el pronóstico visual, realizando el diagnóstico y tratamiento precoz, para evitar complicaciones como la exudación subfoveal crónica y la contracción y fibrosis en la interfase retinovítrea periférica que pueden provocar ectopia macular.
Bibliografía
1. Mukai S, Andreoli CM. Familial exudative vitreoretinopathy. In: Albert DM, Miller JW. Priciples and practice of ophtalmology. Philadelphia: Elsevier; 2008; II: 1865-1870. [ Links ]
2. Ryan SJ. Retina. 4ª Ed. Philadelphia: Elsevier Mosby; 2006; 1-3. [ Links ]
3. Feldman EL, Norris JL, Cleasby GW. Autosomal dominant exudative vitreoretinopathy. Arch Ophthalmol 1983; 101: 1532-1535. [ Links ]
4. Shukla D, Singh J, Sudheer G, Soman M, John RK, Ramasamy K, et al. Familial exudative vitreoretinopathy (FEVR). Clinical profile and management. Indian Ophthalmol 2003; 51: 323-328. [ Links ]
5. van Nouhuys CE. Signs, complications and platelet aggregation in familial exudative vitreoretinopathy. Am J Ophthalmol 1991; 111: 34-41. [ Links ]
6. Criswick VG, Schepens CL. Familial exudative vitreoretinopathy. Am J Ophthalmol 1969; 68: 578-594. [ Links ]
7. Ebert EM, Mukai S. Familial exudative vitreoretinopathy. Int Ophthalmol Clin 1993; 33: 237-247. [ Links ]
8. Pendergast SD, Trese MT. Familial exudative vitreoretinopathy. Results of surgical management. Ophthalmology 1998; 105: 1015-1023. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Ana M. Piñero
Instituto Universitario Barraquer
C/. Laforja, 88
08021 Barcelona
España
E-mail: anapinero@co-barraquer.es
Recibido: 19/3/07.
Aceptado: 10/11/08.

