










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkOncología (Barcelona)
versión impresa ISSN 0378-4835
Oncología (Barc.) vol.29 no.3 mar. 2006
NOTA CLÍNICA
Linfoma pulmonar primario de bajo grado no BALT
F. Ferrer MarínI; M. J. Avilés InglésII
Hospital General Universitario Reina Sofía
Murcia
IUnidad de Hematología
IIUnidad de Neumología
RESUMEN
El linfoma pulmonar primario (LPP) es una patología infrecuente que engloba un amplio espectro de entidades clínico-patológicas, la gran mayoría de las cuales corresponde a linfomas de baja malignidad tipo BALT (linfoma asociado al tejido linfoide bronquial).
Presentamos el caso de una paciente con un linfoma linfocítico B de células pequeñas, primario de pulmón, de bajo grado de malignidad, con la excepcionalidad de no ser encuadrable dentro del subtipo BALT. Discutimos aquí el diagnóstico diferencial histológico entre ambos tipos de linfomas, así como los hallazgos clínico-radiológicos, el pronóstico y la respuesta al tratamiento.
Palabras clave: Linfoma pulmonar primario. Linfoma linfocítico B de células pequeñas. Diagnóstico histológico. Tratamiento.
SUMMARY
Primary lung lymphomas show a broad clinical and cytological spectrum. The most common histological subtypes are low-grade lymphomas from bronchus-associated lymphoid tissue (BALT).
We report a case of non-BALT low-grade lung lymphoma: a primary small B-cell lymphocytic lymphoma. We discuss the histological differential diagnosis, the clinical and radiological findings, prognosis and response to treatment.
Key words: Primary lung lymphoma. Small B-cell lymphocytic lymphoma. Histological diagnosis. Treatment.
Caso clínico
Se trata de una mujer de 72 años con antecedentes de tabaquismo (50 paquetes-año), enolismo y asma extrínseco que consulta por fiebre, tos productiva y aumento de su disnea habitual. En la exploración física presentaba febrícula (37.9º C), taquipnea e hipoventilación con sibilantes espiratorios. No se palpaban visceromegalias ni adenopatías.
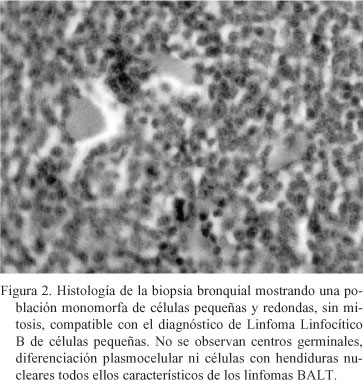
La analítica objetivó una discreta neutrofilia y una trombocitopenia leve y el cultivo y las tinciones de Gram y Ziehl-Neelsen de esputo fueron negativos. La radiografía de tórax (Fig. 1a) mostraba una condensación paracardiaca izquierda, con broncograma aéreo, que borraba la silueta cardiaca y la tomografía axial computerizada (TAC) confirmó la existencia de un infiltrado pulmonar izquierdo, con afectación pleural. Con la sospecha diagnóstica de neumonía de la língula se inició tratamiento con broncodilatadores, oxigenoterapia y amoxicilina-clavulánico con mejoría clínica y desapareción de la febrícula. La enferma fué dada de alta para control en consulta de neumología donde, ante la persistencia de la imagen de condensación, se prosiguió estudio. La broncoscopia evidenció signos inflamatorios inespecíficos en el árbol bronquial izquierdo con normalidad del derecho; la citología del aspirado y cepillado bronquial fué negativa para malignidad. La punción-aspiración con aguja fina (PAAF) guiada por TAC objetivó una proliferación linfoide monomorfa, sospechosa de linfoma no Hodgkin (LNH). Una biopsia pulmonar mediante minitoracotomía confirmó el diagnóstico de Linfoma linfocítico B de células pequeñas (LLBCP) (Fig. 2) CD 20 positivo, bcl-2 positivo, y p-53 negativo.
Los estudios biológicos mostraron un aumento discreto de la VSG, de la b2-microglobulina y la presencia de una banda minimonoclonal IgM lambda en el proteinograma. La serologías víricas y el estudio de autoinmunidad (ANOES) fueron negativos. La ecografía abdominal mostró signos incipientes de hepatopatía crónica difusa y la TAC abdominal no mostraba adenopatías. Una nueva TAC torácica confirmó la presencia de una masa pulmonar densa, homogénea, de 5x7 cm, de forma triangular con base periférica y vértice pericárdico, que afectaba a la língula y el lóbulo superior izquierdo, acompañada de componente atelectásico con desviación del mediastino (Fig. 1c). La biopsia ósea descartó infiltración por linfoma.
Con el diagnóstico de linfoma pulmonar primario (LPP) de bajo grado, se inició tratamiento oral con clorambucilo en pulsos, 40 mg por vía oral día 1, y prednisona, 100 mg/día por 4 días, cada 4 semanas. En la primera reevaluación, tras cuatro ciclos de quimioterapia, el tamaño de la masa tumoral no se había modificado por lo que se propuso a la enferma tratamiento con poliquimioterapia intravenosa que rechazó. Se inició entonces clorambucilo continuo a dosis de 3 mg/día. Una nueva TAC torácica mostró una disminución del diámetro de la masa pulmonar superior al 50% (8 meses después) y mayor del 75% (16 meses después de iniciado el tratamiento diario). Como efectos secundarios presentó toxicidad hematológica y hepática, ambas grado I de la OMS, por lo que se redujo la dosis de clorambucilo a 1 mg/día. Tras dos años de tratamiento continuado, la radiografía (Fig. 1b) y la TAC torácica (Fig.1d) mostraban unos parénquimas pulmonares bien ventilados, apreciándose sólo algún tracto fibroso residual en el pulmón izquierdo (Fig. 1d). Un estudio gammagráfico con galio-67 mostraba hiperactividad a nivel del tercio medio de hemitórax izquierdo, confirmando así la existencia de enfermedad residual. A pesar de ello, la paciente se ha mantenido con abstención terapéutica, en remisión parcial muy buena (RPMB) y libre de progresión hasta la fecha (intervalo libre de progresión de 53 meses).
Discusión
El LPP es una enfermedad infrecuente (menos del 1% de las neoplasias del pulmón y menos del 1% de los LNH)1, que engloba a distintas entidades histológicas y que actualmente se divide en dos grandes grupos anatomoclínicos: a) los LNH de bajo grado (60-90% de los casos), de los que el subtipo asociado al tejido linfoide bronquial (BALT), es con mucho el más frecuente (90% de los de bajo grado) y b) los LPP de alto grado, más raros, y que suelen asociarse a estados de inmunodepresión1, 2. Aunque el diagnóstico diferencial entre estos dos subtipos de LPP sólo puede establecerse sobre bases histológicas, algunos síntomas de presentación y manifestaciones radiológicas difieren entre ambos grupos. Mientras los pacientes con LPP de alto grado casi siempre presentan síntomas generales al diagnóstico, los pacientes con linfomas de bajo grado están asintomáticos y se diagnostican a partir de un hallazgo radiológico casual o bien, como en el caso de nuestra enferma, presentan síntomas respiratorios3. Estos tumores se asocian a enfermedades autoinmunes y a gammapatías monoclonales, generalemente IgM3, como es el caso de nuestra enferma.
Respecto al diagnóstico, hasta en un 50% de los casos la mucosa bronquial tiene un aspecto normal en la broncoscopia y la biopsia es informada como negativa3. Aunque en el caso presentado no se encontró IgM en el sobrenadante del lavado bronquioalveolar (LBA), ésta puede ser encontrada hasta en un 30% de los casos con paraproteínas en el suero4. De hecho, la demostración de una población linfoide B clonal en el LBA se asocia con el diagnóstico de LPP, por lo que su presencia obliga a la realización de procedimientos invasivos, y viceversa5. Finalmente, la PAAF guiada por TAC resulta útil para establecer el diagnóstico de sospecha, aunque la demostración de clonalidad y el diagnóstico histológico preciso requieren una muestra de biopsia de tamaño adecuado. En nuestra enferma, el hallazgo de una masa monomorfa de células linfoides pequeñas y redondas, bcl-2 positivas, y la ausencia de centros germinales, de lesiones linfoepiteliales y de diferenciación plasmocelular, tan características de los linfomas tipo BALT, son compatibles con el diagnóstico histológico de LLBCP y no con el de LPP tipo BALT. Resulta interesante reseñar cómo, en las series más antiguas, el LLBCP era la entidad histológica más frecuente dentro de los LPP de bajo grado6, mientras que en la actualidad ni siquiera se menciona como entidad propia entre los LPP de bajo grado1, 2. Esto es así porque, aunque el concepto del linfoma asociado a mucosas fue introducido en la década de los 807, no es hasta 1994 cuando aparece como una entidad propia en la clasificación de los linfomas8.
Aunque el tratamiento de los LPP de bajo grado sigue siendo controvertido (monitorización, cirugía, monoquimioterapia)1, se puede considerar el clorambucilo oral, a una dosis de 2-4 mg/día, como el tratamiento de elección2,3 , con supervivencias del 100% a los dos años, y superiores al 90% a los cinco años3. La poliquimioterapia se reserva para los linfomas de alto grado o para pacientes con masa bulky2. La eficacia de nuevos fármacos como la fludarabina y el anticuerpo monoclonal anti-CD20 (rituximab) está aún por definir9,10. En nuestra enferma destaca la falta de respuesta al tratamiento con clorambucilo oral en forma de ciclos que contrasta con la magnífica respuesta cuando este mismo fármaco se administró de forma continuada, a diferencia de otros síndromes linfoproliferativos B, como la leucemia linfática crónica, donde se ha demostrado la misma eficacia entre ambas formas de administración11. Posiblemente ésto sea debido a la diferente dosis total recibida según la pauta de administración (40 mg/mes con el esquema de ciclos y 90-60 mg/mes con la pauta diaria).
Aunque nuestra enferma se encuentra en RPMB cinco años después del diagnóstico, continua seguimiento clínico-radiológico, ya que las recaídas, tanto ganglionares como extratorácicas son frecuentes en esta enfermedad3.
Bibliografía
1. Cadranel J, Wislez M, Antoine M. Primary pulmonary lymphoma. Eur Respir J. 2002; 20: 750-62. [ Links ]
2. Habermann TM, Ryu JH, Inwards DJ, Kurtin PJ. Primary Pulmonary Lymphoma. Semin Oncol 1999; 26 (3): 307-15. [ Links ]
3. Chacón JI. Linfoma Pulmonar. En: Garcia-Conde J, Matutes E, Piris MA, Reyes F, editors. Síndrome linfoproliferativos. Madrid, 1999; p. 306-9. [ Links ]
4. Oka M, Kawato K, Kanda T et al. Bronchoalveolarlavage in primary pulmonary lymphoma with monoclonal gammopathy. Am Rev Respir Dis 1986; 134: 590-2. [ Links ]
5. Zompi S, Couderc LJ, Cadranel J et al. Clonality analysis of alveolar B lymphocites contributes to the diagnostic strategy in clinical suspicion of pulmonary lymphoma. Blood 2004, 103: 3208-15. [ Links ]
6. Turner RR, Colby TV, Doggett RS. Well-Differentiated Lymphocytic Lymphoma. A study of 47 patients with primary manifestation in the lung. Cancer 1984; 54: 2088-96. [ Links ]
7. Isaacson P, Wright DH. Extranodal malignant lymphoma arising from mucosa-associated lymphoid tissue. Cancer 1984; 53: 2515-24. [ Links ]
8. Harris NL, Jaffe ES, Stein H, Banks et al. A revised european-american classification of lymphoid neoplasms: a proposal from the international lymphoma study group. Blood 1994; 84: 1361-92. [ Links ]
9. Zinzani PL, Stefoni V, Musuraca G et al. Fludarabine-containing chemotherapy as frontline treatment of non-gastrointestinal mucosa-associated lymphoid tissue lymphoma. Cancer 2004; 100: 2190-4. [ Links ]
10. Raderer M, Jager G, Brugger S et al. Rituximab for treatment of advanced extranodal marginal zone B cell lymphoma of the mucosa-associated lymphoid tissue lymphoma. Oncology 2003; 65: 306-10. [ Links ]
11. Rozman C, Montserrat E. Chronic lymphocitic leukemia. N Engl J Med 1995, 333:1052-7. [ Links ]
 Correspondencia:
Correspondencia:
Dra. F. Ferrer Marín
Unidad de Hematología
C/ S. Martín de Porres, 15 - 2º
E-30001 Murcia
paquiferrer@telefonica.net
Recibido: 31.10.05
Revisado: 01.01.06
Aceptado: 10.02.06

