










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 no.6 Madrid jun. 2004
| POINT OF VIEW |
Hepatitis C and fibrosis
V. Aguilera and M. Berenguer
Service of Digestive Medicine. University Hospital La Fe. Valencia. Spain
Aguilera V, Berenguer M. Hepatitis C and fibrosis. Rev Esp Enferm Dig 2004; 96: 402-414.
Recibido: 28-01-03.
Aceptado: 03-02-04.
Correspondencia: Victoria Aguilera. Servicio de Medicina Digestiva. Hospital Universitario La Fe. Avda. Campanar, 21. 46009 Valencia. Telf.: 96 197 31 18. Fax: 96 197 31 18.
e-mail: mbhaym@teleline.es
INTRODUCTION
Hepatitis C virus (HCV) infection is a health problem involving more than 100 million people worldwide. How-ever, despite these high numbers, only a group of infected people will develop serious difficulties from viral persistence. Liver fibrosis is the main complication of chronic HCV infection, and its eventual outcome -liver cirrhosis- is responsible for liver-related morbidity and mortality (1). The progression rate of fibrosis varies a lot amongst individuals, and its acknowledgment is a determining factor for the assessment of prognosis and treatment needs. Therefore, the search for predictive factors associated with poorer outcome is an interesting work field for the management of these patients.
NATURAL HISTORY OF HEPATITIS C AND ITS PROGRESSION TO FIBROSIS
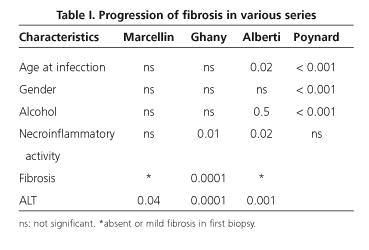
Acute HCV infection results usually in no symptoms and is therefore difficult to diagnose. Overall, it is accepted that only around 15-20% of patients resolve their infection during the acute stage, whereas infection becomes chronic with a persisting viremia in the remaining 80-85% of patients (2). Once established, HCV infection results in liver damage with varying degrees of inflammation and fibrosis in most patients. Even though disease activity is fluctuating in character, the fibrosis stage seems to be both progressive and irreversible (although controversy has currently arisen regarding this statement). Both the outcome and clinical presentation of chronic infection are variable - while some patients progress towards cirrhosis and/or hepatocellular carcinoma, other individuals simply do not develop complications (3,4). Extant studies on the natural history of this disease are heterogeneous regarding both design and population, and no ideal study has been conducted yet (a study including patients with known infection onset time, and long-term treatment-free follow-up). Unfortunately, most studies are cross-sectional with only a single biopsy, and the few longitudinal studies including 2 biopsies are limited by sample size and a relative bias in patient selection. Therefore, the natural history of hepatitis is still controversial and draws much attention at consensus meetings (5-8). A cross-sectional study by Poynard et al, which included 2,235 patients with chronic hepatitis C, assessed fibrosis progression (Table I). The estimated median progression was 0.133 METAVIR units/year, and time needed for the development of cirrhosis was estimated as 30 years, given a linear progression notion is assumed. However, this concept is not currently accepted, and progression is considered to display an asymmetric distribution following at least three progression patterns -rapid, intermediate and slow (9)- and four successive periods for each fibrosis stage since infection (fibrosis progression being slower in early versus advanced stages: 0.056 vs 0.137) (10). Three studies were reported of late, which included two successive biopsies (11-13) and supported this progression model. A systematic review by Freeman et al of 145 studies reporting on the natural history of hepatitis C between 1990 and 2000 included 57 studies in their analysis; they were divided into 4 groups: series of inpatients with chronic liver disease, post-transfusion hepatitis series, cohorts of patients with community-acquired hepatitis, and blood donors. Their major conclusions are summarized as: a) the prevalence of cirrhosis is much higher in inpatients with chronic liver disease and in patients with post-transfusion hepatitis than in patients with community-acquired hepatitis and blood donors (22-24 vs 4-7%); and b) wi-thin 20 years cirrhosis develops in 7% of patients, and this percentage increases to 40% within 40 years, this natural history being much more benign than that previously described by other authors (14). This same team has recently analyzed the studies included in the above-men-tioned review to develop a predictive model for cirrhosis using mathematical calculations, and combining factors such as gender, alcohol ingestion, increased alanine-aminotransferase (ALT) levels, and histologic activity index. This model may prove useful in daily practice when it comes to decide for or against a given therapeutic option (15).
LIVER BIOPSY: ITS CURRENT ROLE
Liver biopsy is still the gold standard in assessing fibrosis stage, and repeat biopsy after 3 or 7 years (time lapse still undefined) is the most effective way to assess fibrosis progression. Various histologic assessment systems have been suggested, among which the HAI or Knodell, modified HAI -which provides a more detailed assessment of fibrosis- and Metavir systems stand out; the latter is most widely used in Europe and is characterized by a lesser degree of intra- and inter-observer variation. However, despite its still relevant role in daily practice, its indications are constantly changing partly due to three factors:
1. Difficulties inherent to its technique.
2. Development of biochemical fibrosis markers or fibrosis predictive models.
3. response variability to antiviral treatments.
1. The first, technical-related difficulty is about the fact that this is an invasive procedure associated with non-negligible morbidity, with major complications in around 0.5% of patients. Secondly, 10-20% of cases show sample errors, since fibrosis is not always homogeneous in the liver, and samples do not always reflect its extension; the presence of cirrhosis may be underestimated in 15-30% of patients. Third, there is strong intra- and inter-oberver variability, with concordance being only 60-90% for fibrosis and less for inflammation (16).
2. Development of non-invasive fibrosis markers: models are currently becoming available that combine serum markers and discriminate between patients with advanced fibrosis (fibrosis bridges or cirrhosis) and mild (< F2) or absent fibrosis. Table II depicts some recent fibrosis predictive models, including their markers (10,17-24). Generally speaking, however, and due to the absence of validation studies with a relevant number of patients, their clinical use is still scarce (21).
3. Finally, response variability to antiviral therapy in selected subgroups will condition the performance or otherwise of a pre-treatment biopsy. Thus, in the subgroup of patients infected by genotypes 2 and 3 the response rate to antiviral therapy using pegylated interferon plus ribavirin is 80%, and therefore a systematic use of biopsy to estimate pre-therapy histological severity is deemed unnecessary. In contrast, biopsy is still an important part of therapeutic strategy for patients with genotypes other than 2 and 3, since therapy may be delayed for patients whose fibrosis is nil or limited to portal tracts when poor virologic response variables (genotype, viral load) are present, since this is a prolonged, side-effect-inducing treatment. In contrast, therapy risks should probably be assumed for patients with advanced fibrosis stages or moderate-severe inflammation regardless of virologic variables. These algorithms will most probably change as antiviral drugs improve.
BIOLOGICAL MARKERS OF FIBROSIS
An alternative approach to liver fibrosis staging is to quantify fibrosis-related products in the serum. Despite the fact that liver fibrosis is a local response, serum levels of fibrogenic citokines, extracellular matrix proteins, and extracellular matrix degradation products may be found in advanced fibrosis stages. Unfortunately, the usefulness of these markers is scarce because of their low sensitivity and specificity. Indeed, one limitation is their lack of organ specificity, as they increase in other clinical conditions. Synthesis and degradation markers include the transforming growth factor ß (TGFß1) -the most significant marker of fibrogenesis involved in hepatic stellate cell activation-, aminoterminal type III procollagen propeptide (PIIINP), and carboxyterminal type III and type IV collagen peptide as collagen matrix degradation products. A mammalian protein (YKL-40) belonging in the chitinase family is being researched of late, as it may prove useful in moderate fibrosis and its progression (25). Amongst extracellular matrix components hyaluronic acid and laminin stand out. In the study by McHutchison serum levels below 60 mi-crograms/L had a negative predictive value of 99% for cirrhosis exclusion (20). Other fibrosis markers include α2-macroglobulin and TIMP-1 [tissue inhibitors of metalloprotease]. Alpha-2-macroglobulin is an acute-phase protein that combined with other molecules may help establish significant fibrosis. TIMP-1 and MMP (matrix metalloproteinase) are extracellular matrix degradation enzymes. A combination of some of these markers, rather than each marker on its own, will probably help predict histologic damage and thus become a replacement of liver biopsy.
FIBROGENESIS
Liver fibrosis results from a biological response to chronic liver damage and subsequent liver remodeling. In patients with chronic HCV infection the liver reacts to viral aggression through an inflammatory response, and this is a component of fibrogenesis development. Initially fibrogenesis is a dynamic process that attempts to repair any external aggression-induced damage, and which is characterized by the synthesis of extracellular matrix molecules, a set of proteins (collagen, elastin), glycoproteins (fibronectin and laminin), and proteoglycans organized and interconnected in a tridimensional network. This synthesis is in turn compensated for by fibrinolysis mechanisms in an attempt to destroy this matrix. This non-organ-specific mechanism is initially intended to limit an ongoing external aggression -hepatitis C virus in this case. However it lingers on and eventually results in misbalance between fibrogenesis and fibrolysis in favor of fibrogenesis, and consequently in extracellular matrix accumulation, which leads to an impairment of liver architecture (26,27). In summary, fibrogenesis is initially beneficial but becomes pathologic upon viral persistence.
In hepatitis C fibrosis starts around portal spaces (periportal fibrosis) and extends onto neighboring spaces and centrolobular veins by way of septa and fibrotic bridges (28-30). The final stage of fibrosis is cirrhosis, which is characterized by the presence of fibrous tissue bands that bring together most portal and centrolobular mesenchymal structures, and isolate liver cell nodules.
Fibrogenesis is directly related to hepatic stellate cell activation (31-34). In a healthy liver these cells are found in Disse's space between hepatocytes and the sinusoid wall, and make up 5-8% of all cells. Their function is retinoid (vitamin A) storage. In the presence of liver damage they undergo activation (transdifferentiation) and acquire a phenotype similar to that of fibroblasts, with a contractile cytoskeleton characterized by the expression of smooth muscle alpha-actin; they see to the production and remodeling of extracellular matrix (34).
Various factors have been involved in liver fibrogenesis, among which the following stand out: cytokines, chemokynes (a family of cytokines capable of inducing cell migration), extracellular matrix receptors, adhesion molecules (ICAM-1, N-CAM), endotelin, norepinephrine, metalloproteinases, metalloproteinase inhibitors, and growth factors. A most relevant growth factor that plays a role in hepatic stellate cell activation is TGF-ß. PDGF (platelet-derived growth factor) is also a powerful mitogen for hepatic stellate cells. IL-13 and angiotensin II are both significant cytokines involved in stellate cell activation. CTGF has also been documented in cases of hepatitis C virus infection, and various techniques have shown CTGF increases (33-38) that overall correlate to liver fibrosis (35). Other molecules that play a role in the process of fibrogenesis are those involved in stellate cell migration to damage sites, as is the case with endotelin. IL-10 has been involved in the process of hepatic stellate cell contraction. The activation of ECM-destructive proteinases has been related to γ-interferon. Other molecules that also seem to play a role in hepatic stellate cell activation include lipid peroxidation by-products (37). Amongst chemokynes, CCL21 is implied in fibrogenesis-promoting T-cell recruitment (39). MCP-1 is one of the most potent molecules in recruiting monocytes and macrophages (40). The role of autonomic nervous system cytokines and receptors in fibrogenesis development is a major area currently under study.
Of all these factors, the most clinically relevant ones include: a) those related to fibrogenesis: IL-13, angiotensin II, TGF-ß, and γ-interferon; and b) those related to fibrinolysis: interleukin-10.
In summary, fibrogenesis results in the development of fibrosis and a restructuring of the extracellular matrix within hepatic cell tissue, which leads to the lax ECM becoming a reticulated, dense, fibrillar ECM that is more resistant to enzymatic degradation.
FACTORS ASSOCIATED WITH FIBROSIS DEVELOPMENT IN CHRONIC HEPATITIS C
Cross-sectional and longitudinal epidemiologic studies allowed to define clinical factors associated with the rate of fibrosis development in hepatitis C. These factors may be virus-, host-, and environment-related (Table III).
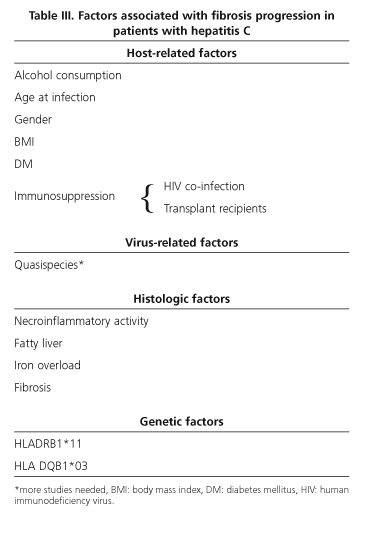
Regarding viral factors, several studies initially suggested an association between high viral load or genotype 1 infection and aggressive disease. Both these associations have been seemingly ruled out. Regarding viral load, most cross-sectional studies found no correlation between high viremia and greater activity (38). In fact, patients with normal transaminase levels and scant histological activity in biopsy samples may have high viremias. Regarding genotype, recent studies did not confirm the suggested association between genotype 1 and cirrhosis or hepatocellular carcinoma outcomes. The longer disease by genotype 1 versus non-1 probably explains differences amongst studies (41). High viral mutagenicity and its complex composition regarding quasispecies seem to allow this virus to escape the host's immune surveillance system, thus facilitating disease progression. Quasispecies heterogeneity has also been related to greater liver damage (42), and less variability has been seen in patients whose infection resolves, while a high genetic variability exists in cases that evolve towards severe chronic hepatitis. Further studies are needed to define the role of quasispecies in the natural history of hepatitis C.
Host-related factors such as age at infection and gender seem to be most significant for hepatitis progression. Thus, infection at later ages is seemingly associated with more aggressive disease. Studies in patients with post-transfusion hepatitis suggest that at least 20% of subjects that become infected at 40 or more will develop cirrhosis within 15 years (43,44). However, only 5% of patients infected via immunoglobulins at younger ages will develop cirrhosis within 20 years following infection (45,46). Similarly, fibrosis progression from 50 years on has been demonstrated to be greater regardless of age at infection (9,10). Finally, donor age in liver transplant recipients seems to play a relevant role in the natural history of recurrent hepatitis C. Male sex has also been associated with poorer outcome in most studies performed. This fact would account for the milder outcomes seen in young women. The reason is un-known. Confounding factors such as body mass index (BMI), alcohol consumption or age at infection may justify this. The host's immune status clearly influences the natural history of hepatitis C. Thus, progression towards cirrhosis is greater in HIV-coinfected patients and in liver transplant recipients with recurrent hepatitis C. In parenteral substance abusers a progression to cirrhosis within 7 years has been described for HIV-positive individuals versus 23 years for HIV-negative individuals (47,48). In patients transplanted for cirrhosis secondary to HCV infection, graft cirrhosis develops in 30% of subjects at 5 years, and the median time for the development of this cirrhosis is only 12 years (49,50). A greater progression of fibrosis has also been seen in newly transplanted patients. The causes of such worsen-ing have not been elucidated yet, but donor older age and more powerful immunosuppression may likely play a relevant role (51).
Regarding environmental factors, alcohol consumption above 50 g per day favors greater fibrosis progression, particularly from 10 years after infection on. Even small amounts have been seen to result in greater fibrosis progression (9,10,52-54).
External factors notwithstanding, other factors such as histological factors are associated with greater fibrosis progression, and are only acknowledged after a biopsy is performed. These include necroinflammatory lesions, steatosis, iron overload and fibrosis. Necroinflammatory activity is a dynamic process that fluctuates over time. Its presence and severity in a biopsy sample seem to be related to eventual fibrosis outcome (12,13). Steatosis is a common finding in hepatitis C biopsies, more common in fact in those infected by genotype 3 and in patients with a high BMI or diabetes mellitus (55,56). Although its pa-thophysiologic mechanism remains unknown, an association between steatosis severity and fibrosis and/or fibrosis progression has been seen. Iron overload is common though moderate in patients with hepatitis C. Its role in the progression of fibrosis is debatable. It seems to be associated with grater inflammatory activity or high alcohol consumption. Results from clinical studies suggesting that iron overload favors poorer fibrosis outcomes are contradictory (58). Lastly, in patients undergoing several biopsies a predictive factor for fibrosis or cirrhosis development is the presence of previous fibrosis, since fibrosis itself may activate fibrogenesis (59,60).
In addition to virus-, host-, environment-, and histology-related factors, individual genetic susceptibility may play an important role regarding the outcome of this disease (61). Thus, it has been reported that alleles HLA DRB1*11 and HLA-DQB1*03 favor viral clearance and associate with a lower risk of liver lesion development (61-63).
The presence of various polymorphisms in the genes cod-ing for immunoregulating proteins, cytokines or fibrogenic factors is probably important for fibrosis outcome both in patients with chronic hepatitis C and patients with alcohol-related liver disease or primary biliary cirrhosis (64).
IS FIBROSIS OR CIRRHOSIS REVERSAL POSSIBLE IN HEPATITIS C?
The notion of fibrosis or cirrhosis reversibility is not new. Currently, clinical trials on the treatment of HCV infection have renewed interest in this concept, and are favoring a change in the paradigm of hepatology. Experimental animal models of early liver damage have shown that the regression of fibrosis or cirrhosis is feasible as a result of interstitial collagenase or tissue inhibitors of metalloproteinase destroying the extracellular matrix (as a result of hepatic stellate cell activation), as well as the activation of hepatic stellate cell apoptosis (65,66). Fibrosis regression has also been reported in humans, but available studies are few and include diverse liver disease etiologies. In all of them fibrosis/cirrhosis reversal was achieved after the cause of liver disease was eliminated. More than 30 years ago cirrhosis reversibility was already reported in patients with hemocromatosis that had undergone therapy using phlebotomies, as well as in patients with Wilson's disease following treatment with D-penicillamine (67,68). Fibrosis improvement was also described in patients with autoimmune hepatitis and primary biliary cirrhosis following immunosuppressive therapy (69-71), and evidence exists regarding fibrosis reversibility in patients with chronic hepatitis B and D following a regimen of lamivudine (72) and interferon, respectively. Cirrhosis reversibility has been recently described following biliary decompression in patients with chronic pancreatitis and main bile duct stenosis (73).
Whether available antiviral therapies may influence the progression of fibrosis and even lead to fibrosis reversal has been pondered for long regarding patients with HCV infection. The immunomodulating effects of interferon may help reduce fibrosis (74,75). In addition, acting on the various factors related to worse infection progression -such as a high BMI- also seems to result in improved liver fibrosis and steatosis, as well as in a reduction of hepatic stellate cells (76). In patients with iron overload and HCV infection serial phelobotomies are also associated with biochemical and histologic improvement (77,78). Poynard et al recently described a reduction of fibrosis and even cirrhosis reversal in patients treated with pegylated interferon plus ribavirin. This fact seems to take place particularly in patients who achieve viral clearance, who have a BMI below 27%, and who are younger than 40 years. These findings suggest that complete reversibility may be obtained for newly establi-shed or "young" cirrhosis (79).
In summary, it is possible to revert or reduce fibrosis using antiviral therapy and/or acting on factors associated with greater disease aggressiveness. Further research is needed on the molecular mechanisms of fibrosis regression using antifibrosing agents. However, a complete arrest of fibrosis progression will only be achieved through hepatitis C virus elimination. It is necessary that better antiviral drugs be developed in order to systematically allow this to happen.
ACKNOWLEDGEMENTS
This paper was partly supported by a grant from Instituto de Salud Carlos III (C03/02).
REFERENCES
1. Khan MH, Farell GC, Bit K, Lin R, Weltman M, George J, et al. Wich patients with hepatitis C develop liver complications? Hepatology 2000; 31: 513-20.
2. Orland JR, Wrigth TL, Cooper S. Acute hepatitis C. Hepatology 2001; 33: 321-7.
3. Pagliaro L, Peri V, Linea C, Cama C, Giunta M, Magrin S. Natural history of chronic hepatitis C. Ital J Gastroenterol 1999; 31: 28-44.
4. Alter HJ, Seeff LB. Recovery, persistence and sequelae inhepatitis C virus infeccion: a perspective on long term outcome. Semin Liver Dis 2000; 20: 17-35.
5. Seef LB. Natural history of hepatitis C. Hepatology 1997; 26 (Supl. 1): 21S-28S.
6. Liang TJ, Rehermann B, Seef LB, Hoofnagle JH. Pathoghenesis, natural history, treatment and prevention of hepatitis C (NIH Conference). Ann Intern Med 2000; 132: 296-305.
7. Alberti A, Chemello L, Benbegnù L. Natural history of hepatitis C. J Hepatol 1999; 31(supl. 1): 17-24.
8. Seef LB. Natural history of chronic hepatitis C. Manegement of hepatitis C: 2002 (NIH Consensus Statement). //consensus nih.gov/116/116cdc_intro-htm.
9. Poynard T, Bedossa P, Opolon P, for the OBSVIRC, CLINIVIRC and DOSVIRC groups. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet 1997; 349: 825-32.
10. Poynard T, Ratziu V, Charlotte F, Goodman Z, McHutchison J, Albrecht J. Rates and risks factors of liver progression in patients with chronic hepatitis C. J Hepatol 2001; 34: 730-9.
11. Marcellin P, Akrémi R, Cazals D, Boyer N, Aupérin A, Vidaud D, et al. Genotype 1 is associated with a slower progression of fibrosis in untreated patients with mild chronic hepatitis C. J Hepatol 2001; 34(suppl. 1): 159A.
12. Ghany MG, Kleiner DE, Alter HJ, Doo E, Khokhar F, Park Y, et al. Progression of fibrosis in early stages of chronic hepatitis C. Hepatology 2000; 32: 496A.
13. Alberti A, Boccato S, Ferrari A, Benbegnu L, Pontisso P, Noventa F, et al. Outcome of initially mild chronic hepatitis C Hepatology 2001; 34: 225A.
14. Freeman AJ, Dore GJ, Law MG, Thorpe M, Von Oberbeck J, Lloyd R, et al. Estimating progression to chirrosis in chronic hepatitis C virus infection. Hepatology 2001; 34: 809-16.
16. Fontana RJ, Lok AS. Non-invasive monitoring of patients with chronic hepatitis C. Hepatology 2002; 36: S57-S64.
17. Guechot J, Laudat A, Loria A, Serfaty L, Poupon R, Giboudeau J. Diagnostic accuracy of hyaluronan and type III procollagen amino-terminal peptide serum assays as markers of liver fibrosis in chronic viral hepatitis C evaluated by ROC curve analysis. Clin Chem 1996; 42: 558-63.
18. Oberti F, Valsesia E, Pilette C, Rousselet MC, Bedossa P, Aube C, et al. Non-invasive diagnosis of hepatic fibrosis or cirrhosis. Gastroenterology 1997; 113: 1609-16.
19. Bonacini M, Hadi G, Govindarajan S, Lindsay KL. Utility of a discriminant score for diagnosing advanced fibrosis or cirrhosis in patients with chronic hepatitis C virus infection. Am J Gastroenterol 1997; 92: 1302-4.
20. McHutchison JG, Baltt LM, de Medina M, Craig JR, Conrad A, Schiff ER, et al. Mesurement of serum hyaluronic acid in patients with chronic hepatitis C and its relationship to liver histology. Consensus Interferon Study Group. J Gastroenterol Hepatol 2000; 15: 945-51.
21. Imbert-Bismut F, Ratziu V, Pieroni L, Charlotte F, Benhamou Y, Poynard T. Multiviric group. Biochemical markers of liver fibrosis in patients with hepatitis C virus infection: a prospective study. Lancet 2001; 357: 1069-75.
22. Patel K, Gordon SC, Smith K, et al. A non-invasive panel of serum markers can reliably differentiate hepatitis C patients with minimal fibrosis from those with fibrosis stages F2-F4. Hepatology 2002; 36: 355A
23. Forns X, Ampurdanes S, Llovet JM, Aponte J, Guinto L, Martínez Bauer E, et al. Identification of chronic hepatitis C patients without hepatic fibrosis by a simple predictive model. Hepatology 2002; 36: 986-92.
24. Chun-Tao-Wai, Greenson JK, Fontana RJ, Kalbfleisch JD, Marrero JA, Conjeevaram HS, et al. A simple non-invasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003; 38: 518-26.
25. Johansen JS, Christoffersen P, Moller S, et al. Serum YKL-40 is increased in patients with hepatic fibrosis. J Hepatol 2000; 32: 911-20.
26. Schuppan D. Structure of the extracellular matrix in normal and fibrotic liver: Collagens and glicoproteins. Semin Liver Dis 1990; 10: 1-10.
27. Martínez Hernández A, Amenta PS. Morphology, localization and origin of the hepatic extracellular matrix. In: Zem MA, Reid LM, eds. Extracellular matrix, chemistry, biology and pathobiology with emphasis on the liver. New York: M Dekker, 1993. p. 201-54.
28. Lefkowitch JH, Schiff ER, Davis GL, Perrillo RP, Lindsay K, Bodenheimer HC, et al. Pathological diagnosis of chronic hepatitis C. A multicenter comparative study with chronic hepatitis B. Gastroenterology 1993; 104: 595-603.
29. Scheuer PJ, Ashrafzadeh P, Sherlock S, Brown D, Dusheiko GM. The pathology of hepatitis C. Hepatology 1992; 15: 567-71.
30. Goodman ZD, Ishak. KG. Histopathology of hepatitis C virus infection. Semin Liver Dis 1995; 15: 70-81.
31. Eng FJ, Friedman SL. Fibrogenesis I. New insights into hepatic stellate cell activation: the simple becomes complex. Am J Physiol 2000; 279: G7-G11.
32. Friedman SL, Roll FJ. Isolation and culture of hepatic lipocytes, Kuffer cells and sinusoidal endothelial cells by density gradient centrifugation with Stracant. Anal Biochem 1987; 161: 207-18.
33. Bedossa P. The cell origin of extracellular matrix proteins. J Hepatol 1993; 19: 1-3.
34. Areson DM, Friedman SL, Bissel DM. Formation of extracellular matrix in normal rat liver: lipocytes as a mayor source of proteoglycan. Gastroenterology 1998; 95: 441-7.
35. Paradis V, Dargere D, Vidaud M, De Gouville AC, Huet S, Martínez V, et al. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology 1999; 30: 968-76.
36. Bedossa P, Poynard T, Mathurin P, Lemaigre ,G, Chaput JC. TGF-beta 1 in situ expression in the liver of patients with chronic hepatitis C treated with alpha interferon. Gut 1993; 34 (Supl. 2): S146-S147.
37. Paradis V, Mathurin P, Kollinger M, Imbert-Bismut F, Charlotte F, Piton A, et al. In situ detection of lipid peroxidation products in chronic hepatitis C: correlations with clinical features. J Clin Pathol 1997: 50: 401-7.
38. Zeuzem S, Franka A, Lee JH, Herrmann G, Rüster B, Roth WK. Phylogenetic analysis of hepatitis C virus islotates and their correlations to viremia, liver function tests and histology. Hepatolgy 1996; 24: 1003-9.
39. Marra F. Hepatic stellate cells and the regulation of liver inflammation. J Hepatol 1999; 31: 1120-30.
40. Boring L, Charo IF, Rollings BJ. MCP-1 in human disease: insights gained from animal models. In: Totowa N, ed. Chemokines in disease: biology and clinical research. 1st ed. Totowa, NJ: Humana, 1999. p. 53-65.
41. López-Labrador FX, Ampurdanes S, Forns X, Castells A, Saiz JC, Costa J, et al. Hepatitis C virus (HCV) genotypes in Spanish patients with HCV infection: relationship between HCV genotype 1b, cirrhosis and hepatocellular carcinoma. J Hepatol 1997; 27: 959-65.
42. Pawlotsky JM, Pellerin M, Bouvier M, Roudot Thoraval F, Germanidis G, Bastie A, et al. Genetic complexity of the hypervariable region 1 (HVR 1) of hepatitis C virus. Influences on the characteristics of the infection and the response to alpha-interferon therapy in patients with chronic hepatitis C. J Med Virol 1998; 54: 256-64.
43. Alter HJ, Seef LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective in long-term outcome. Semin Liver Dis 2000; 20: 17-35.
44. Tong MJ, El-Farra NS, Reijes AR, Co RL. Clinical outcomes after transfusion-associated hepatitis C. N Engl J Med 1995; 332: 1463-6.
45. Kenny-Walsh E. Clinical outcomes after hepatitis C infection from contamined anti-D immune globuline. N Engl J Med 1999; 340: 1228-33.
46. Wiese M, Berr F, Lafrenz M, Porst H, Oesen U. Low frequency of cirrhosis in a hepatitis C (genotipe 1b) single -source outbreak in Germany: a 20 years multicenter study.Hepatology 2000; 32: 91-6.
47. Soto B, Sanchez-Quijano A, Rodrigo L, Leal M, Lissen E. HIV infection modifies the natural history of chronic parenterally adquired hepatitis C with an unusually rapid progresión to cirrosis. A multicenter study on 547 patients. J Hepatol 1997; 26: 1-5.
48. Di Martino V, Rufat P, Boyer N, Renard P, Degos F, Martinot-Peignoux M, Matheron S, et al. Influence of human immunodeficiency virus coinfection on chronic hepatitis C in injection drug users: a long term retrospective cohort study. Hepatol 2001; 34: 1193-9.
49. Prieto M, Berenguer M, Rayon M, Cordoba J, Argüello L, Carrasco D, et al. High incidence of allograft cirrosis in hepatitis C virus genotipe 1b infection following transplantation: relationship with rejections episodes. Hepatology 1999; 29: 250-6.
50. Berenguer M, Ferrel L, Watson J, Prieto M, Kim M, Rayon M, et al. HCV-related fibrosis progression following liver transplantation: increase in recent years. J Hepatol 2000; 32; 673-84.
51. Berenguer M, Prieto M, San Juan F, Rayón JM, Martínez F, Carrasco D, et al. Contribution of donor age to the recent decrease in patient survival among HVC-infected liver transplant recipients. Hepatology 2002; 36: 202-10.
52. Wiley TE, Mc Carthy M, Breidi L, Mc Carthy M, Layden TJ. Impact of alcohol on the histological and clinical progression of hepatitis C infection. Hepatology 1998; 28: 805-9.
53. Pessione F, Degós F, Marcellin P, Duchatelle V, Njapoum C, Martinot-Peignoux M, et al. Effect of alcohol consumption on serum hepatitis C virus RNA and hitological lesions in chronic hepatitis C. Hepatology 1998; 27: 1717-22.
54. Khan KN, Yatsuhasi H. Effect of alcohol consumption on the progression of hepatitis C virus infection and riscks of hepatocellular carcinoma in Japanese patients. Alcohol Alcohol 2000; 35: 286-95.
55. Adinolfi LE, Gambardella M, Andreana A, Tripodi M-F, Utili R, Ruggiero G. Steatosis accelerates the progresión of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 2001; 133: 1358-64.
56. Ortiz V, Berenguer M, Rayon JM, Carrasco D, Berenguer J. Contribution of obesity to hepatitis C-related fibrosis progresión. Am J Gastroenterol 2002; 97: 2408-14.
57. Monto A, Alonzo J, Watson J, Grunfeld, Wrigth TL. Steatosis in chronic hepatitis C: Relative contributions of obesity, diabetes mellitus and alcohol. Hepatology 2002; 36: 729-36.
58. Piperno A, Vergani A, Malosio I, Parma L, Fossati L, Ricci A, et al. Hepatic iron overload in patients with chronic viral hepatitis: role of HFE gene mutations. Hepatology 1998; 28: 1105-9.
58. Piperno A, Vergani A, Malosio I, Parma L, Fossati L, Ricci A, et al. Hepatic iron overload in patients with chronic viral hepatitis: role of HFE gene mutations.59. Paradis V, Mathurin P, Charlotte F, Vidaud M, Pooynard T, Hoang C, et al. Histological features predictive of fibrosis in chronic hepatitis C infection. J Clin Pathol 1996; 49: 1-7.
60. Yano M, Kumada H, Kage M, Ikeda K, Shimamatsu K, Inoue O, et al. The long-term pathological evolution of chronic hepatitis C. Hepa-tology 1996; 23: 1334-40.
61. Thursz M. Genetic susceptibility in chronic viral hepatitis. Antivir Res 2001; 52: 113-6.
62. Mangia A, Gentile R, Cascavilla I, Margaglione M, Villani MR, Ste-lla F, et al. HLA class II favors clearance of HCV infection and pro-gression of the chronic liver damage. J Hepatol 1999; 30: 984-9.
63. Tillmann HL, Chen DF, Trautwein C, Kliem V, Grundey A, Berning-Haag A, et al. Low frequency of HLA-DRB1*11 in hepatitis C virus induced end stage liver disease. Gut 2001; 48: 714-8.
64. Bataller R, North KE, Brenner DA. Genetic polymorphisms and the progression of liver fibrosis: a critical appraisal. Hepatology 2003; 37: 493-503.
65. Mühlbauer M, Bosserhoff A, Hartmann A, Thasler W, Weiss T, Her-farth H. A novel MCP-1 Gene polymorphism is associated with hepa-tic MCP-1 expression and severity HCV-related liver disease. Gastro-enterol 2003; 125: 1058-93.
66. Issa R, Williams E, Trim N, Kendall T, MJP Athur, Reichen J, et al. Apoptosis of hepatic stellate cells: involvement in resolution of bi-liary fibrosis and regulation by soluble growth factors. Gut 2001; 48: 548-57.
67. Powell LW, Kerr JF. Reversal of long term intensive venesection therapy. Australs Ann Med 1970; 19: 54-7.
68. Falkmer S, Samuelson G, Sjölin S. Penicillamide –induced normali-zation of clinical signs and liver morphology and histochemistry in a case of Wilson's disease. Pediatrics. 1970: 45: 260-8.
69. Dufour JF, DeLellis R, Kaplan MM. Reversibility of hepatic fibrosis in autoimmune hepatitis. Ann Intern Med 1997; 19: 54-7.
70. Wanless IR. Use of corticosteroid therapy in autoimmune hepatitis in the resolution of cirrhosis. J Clin Gastroenterol 2001; 32: 371-2.
71. Kaplan MM, Delellis RA, Wolfe HJ. Sustained biochemical and his-tologic remission of primary biliary cirrhosis in response to medical treatment. Ann Intern Med 1997; 126: 682-8.
72. Kweon YO, Goodman ZD, Dienstag JL, Schiff ER, BrownNA, Burk-hardt E, et al. Decreasing fibrogenesis: an immunohistochemical study of paired liver biopsies following lamivudine therapy of chronic hepatitis B. Br J Hepatol 2001; 35: 749-55.
73. Hammel P, Couvelard A, OToole D, Ratouis A, Sauvanet A, Flejou JF, et al. Regression of liver fibrosis after biliary drainage in patients with chronic pancreatitis and stenosis of the common bile duct. N Engl J Med 2001; 344: 418-23.
74. Sobesky R, Mathurin P, Charlotte F, Moussali J, Olivi M, Vidaud M, et al. Modeling the impact of interferon alfa treatment on liver fibro-sis progression in chronic hepatitis C: a dynamic view. Gastroentero-logy 1999; 116: 378-86.
75. Shiratori Y, Imazeki F, Moriyama M, Yano M, Arakawa Y, Yokosu-ka O, et al. Histologic improvement of fibrosis in patients with hepati-tis C who have sustained response to interferon therapy. Ann Intern Med 2000; 132: 517-24.
76. Hickman IJ, Clouston AD, Macdonald GA, Purdie DM, Prins JB, Ash S, et al. Effect of weight reduction on liver histology and bioche-mistry in patients with chronic hepatitis C. Gut 2002; 51: 89-94.
77. Fargion S, Fracanzani AL, Rossini A, Borzio M, Riggio O, Belloni G, et al. Iron reduction and sustained response to interferon-alpha ther-apy in patients with chronic hepatitis C: results of an Italian multicen-ter randomized study. Am J Gastroenterol 2002; 97: 1204-10.
78. Yano M, Hayashi H, Wakusawa S, Sanae F, Takikawa T, Shiono Y, et al. Long term effects of phlebotomy on biochemical and histological parameters of chronic hepatitis C. Am J Gastroenterol 2002 ; 97: 133-7.
79. Poynard T, McHutchison J, Trepo C, Lindsay K, Goodman Z, Mei-Hsiu L, et al. Impact of pegylated Interferon ala-2-b and ribavirine on liver fibrosis in patients with chronic hepatitis C. Gastroenterology 2002; 122: 1303-13.


{kind=link}