










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versão impressa ISSN 1130-0108
Rev. esp. enferm. dig. vol.97 no.8 Madrid Ago. 2005
| POINT OF VIEW |
IL-6 and extracellular matrix remodeling
J. A. Solís Herruzo, P. de la Torre, T. Díaz Sanjuán, I. García Ruiz and T. Muñoz Yagüe
Department of Medicine, Gastroenterology. Research Center. Hospital Universitario 12 de Octubre. Madrid, Spain
Solís Herruzo JA, de la Torre P, Díaz Sanjuán T, García Ruiz I, Muñoz Yagüe T. IL-6 and extracellular matrix remodeling. Rev Esp Enferm Dig 2005; 97: 575-595.
Acknowledgment: This study was supported in part by a Grant for Medical Research from the "Fundación Mutua Madrileña" (Spain, 2004).
Recibido: 04-01-05.
Aceptado: 08-01-05.
Correspondencia: José A. Solís Herruzo. Servicio de Patología Digestiva. Hospital 12 de Octubre. Avda. de Córdoba, s/n. 28041 Madrid. Fax: 91 3908280.
e-mail: jsolis.hdoc@salud.madrid.org
ABBREVIATIONS
AP1: activator protein 1; CAT: Chloramphenicol acetyl transferase; ECM: Extracellular matrix; HSCs: Hepatic stellate cells; Jak: Janus tyrosine kinases; JNK: c-Jun N-terminal kinase; MMPs: Matrix metalloproteinases; PP2A: Protein phosphatase 2A; TIMPs: Tissue inhibitor MMPs; IL-6: Interleukin-6.
INTRODUCTION
Hepatic fibrosis is a characteristic feature of chronic liver diseases. It is the result from the excessive deposition of extracellular matrix (ECM) proteins into the liver in response to a number of liver injuries, including chronic ethanol ingestion, viral hepatitis and iron deposition. Hepatic fibrosis has important physiopathologic consequences on the liver function and structure. First, it may contribute to increase intrahepatic vascular resistances to the portal flow and to originate portal hypertension. Activated hepatic stellate cells may contribute to this effect, since these cells exhibit contractile capacity and can modulate blood flow at the sinusoidal level (1). Second, perisinusoidal fibrosis represents a barrier to the exchange of nutrients and metabolites between sinusoidal blood and hepatocytes. Furthermore, deposition of ECM in the space of Disse induces the loss of fenestrae of the sinusoidal endothelial cells (2). Third, ECM is a physiological reservoir for cytokines and growth factors in the close cell surrounding. The binding of these factors to ECM component modulates their biological activity and protects them from proteolysis (3).
EMC of the liver is composed of three distinct types of proteins including collagen, noncollagenous glycoproteins, and proteoglycans (Fig. 1). Collagen type I, III and V are heterotrimer molecules that form a triple helical structure with a tendency to form supramolecular aggregates (4,5). Type V, a pericellular collagen in the normal liver, is also present in increased amount in septa and portal tracts. Collagen IV is the major collagen of basement membrane. It is an unusual collagen type in that molecules cross-link at their terminal domains to form a network. This sheet of collagen is physically intermeshed or directly bound to other components of the basement membrane like laminins, nidogen, or perlecan.
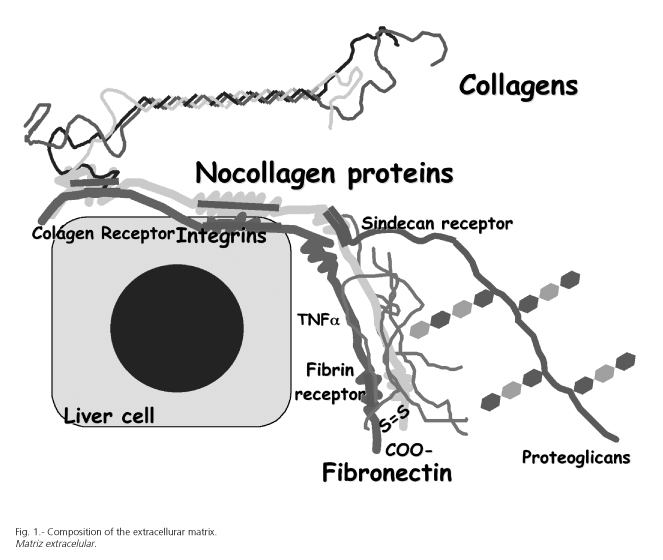
In the hepatic fibrosis and cirrhosis, all types of collagens are increased. When hepatic fibrosis is mild, collagen type I and III are equally increased. However, when hepatic fibrosis is severe, the predominant collagen in the connective tissue bands is type I, and the ratio of collagen I to collagen III increased to 4:1 (5). In these conditions, collagen IV is deposited mainly in the space of Disse, transforming sinusoids into capillaries. Although collagen I becomes the predominant ECM constituent in fibrosis and cirrhosis, many other proteins also are part of the fibrous complex. Thus, glycoproteins (fibronectin, laminins, tenascin, undulin, thrombospondin, SPARC) and proteoaminoglycans (perlecan, biglycan, decorin, aggrecan) also are increased in scars and septa.
Although it is likely that each cell population in the liver plays a role in the production of ECM components, hepatic stellate cells (HSCs) appear to be the major fibrogenic cell type (6). HSCs are resident nonparenchymal cells located in the subendothelial space between hepatocytes and sinusoidal endothelial cells. In normal liver, HSCs are relatively quiescent with respect to ECM synthesis (7,8). However, during liver injury, they undergo a complex process that transforms quiescent HSC to one activated cell that is proliferative, fibrogenic, and contractile (9,10). HSC activation is associated with changes in the metabolism (increased DNA synthesis, cellular proliferation), the pattern of gene expression [collagen I, III, IV, laminin, fibronectin, MMPs (Matrix Metalloproteinases), TIMPs (Tissue Inhibitor of MMPs)], and the capacity to synthesize cytokines, growth factors and their receptors.
Extensive fibrosis in experimental liver injury may resolve upon withdrawal of the injurious agent. Thus, fibrosis or cirrhosis induced by biliary obstruction or carbon tetrachloride intoxication in rat is reversible over a period of one to three months (11). Likewise, reversible fibrosis has been shown in mouse and rabbit models of hepatosplenic schistosomiasis (12). Current evidence indicates that regression of liver fibrosis and advanced micronodular cirrhosis is a real phenomenon if the original cause of the liver injury is effectively removed (13-17). However, this process may be incomplete over a prolonged period and results in the development of maconodular cirrhosis. Remodeling of this residual lesion is limited by matrix cross-linking (18). Transglutaminase-mediated cross-linking of collagen provides matrix proteins with resistance to MMP-mediated degradadation (19). Thus, these linking may limit matrix degradation even in the presence of active MMPs.
Degradation of the ECM in the hepatic fibrosis is a complex process involving MMPs, specific TIMPs, and enzymes that activate latent MMPs (20). In an experimental model of liver fibrosis, transient MMPs overexpression in the liver effectively attenuates established fibrosis (21).
MMPs constitute a large family of structurally related zinc-dependent endopeptidases capable of degrading a wide variety of ECM components (22-24), and, therefore, they may play a key role in the resolution of liver fibrosis upon withdrawal of the injurious agent. MMP-1 and MMP-13 cleave interstitial collagens (types I, II, III and X). MMP-2 (gelatinase A) and MMP-9 (gelatinase B) degrade denatured interstitial collagen (gelatins), type V and IV collagens, and noncollagen proteins (fibronectin, laminin, elastin). MMP-3 (stromelysin-1), MMP-10 (stromelysin-2) and MMP-7 (matrilysin) degrade a broad range of substratres, including proteoglycans, and collagen and noncollagen proteins. Membrane-type MMPs (MMP-14 to MMP-25) are anchored to the surface of cells through their carboxyl-terminal transmembrane domain. These membrane-type MMPs degrade collagen and noncolagenous proteins, proteoglycans and play an important role in the activation of MMP-2 and MMP-13. In rats and mice, there is only one interstitial MMP, the MMP-13, which shares 86% homology with human MMP-13. MMP-13 is capable of degrading type I, II, III, IV, X, and XIV collagens, tenascin, fibronectina and the aggrecan protein (25).
MMPs are secreted as inactive proenzymes; therefore they need to be subsequently activated by several mechanisms including enzymatic cleavage (serine protease plasmin, stromelysin-1, membrane-type MMPs) (20,26). Activated MMPs produce a limited number of cleavages within the triple helix of the three a-chain of interstitial collagen molecules, e.g. in type I collagen, cleavage occurs between Gly775 and Ile776 in the a1 chain and the corresponding Gly/Leu in the a2 chain, to yield typical three-fourths length amino-terminal and one-fourth length carboxyl-terminal fragments. This initial single site cleavage of interstitial collagens results in their partial unfolding, which then renders them susceptible to degradation to small fragments by gelatinases. The activity of MMPs is regulated by a complex mechanism at the level of gene expression, proenzyme activation, or binding of proenzyme or active enzyme to TIMPs.
TIMPs are low molecular weight glycoproteins capable of irreversibly binding to proenzyme or active form of MMPs with a 1:1 stoichiometry and inactivating a wide variety of MMPs degrading enzymes. Because of the one-to-one interactions, small changes in levels of TIMPs can lead to significant changes in the biological activity of MMPs. Excess production of TIMPs relative to MMPs could contribute to the progression of liver fibrosis, while the enhancement of matrix breakdown may be achieved by either reducing the synthesis of TIMPs or increasing the expression of MMPs. All members of this family are able to inhibit all of the MMPs. However, some TIMPs have specific relationship with individual MMPs (27,28). TIMPs are multifunctional molecules that not only inhibit MMPs but also stimulate proliferation of various cell types and can protect cells from apoptosis (29,30).
The severity of hepatic fibrosis is the result of the degree of collagen synthesis and the fate of the collagen deposited into the ECM of the liver, which, in turn, depend on the balance between the production and the activation of MMPs and the levels of TIMPs. In chronic liver disease in human and in animal models of liver fibrosis, expression of MMP-1/MMP-13 does not change significantly, as hepatic fibrosis develops and progresses, but there is a striking increase in expression of TIMP-1 and TIMP-2 (31). In homogenates prepared from fibrotic livers, the level of TIMP-1 protein increases approximately five-fold in comparison with normal liver. Regression of hepatic fibrosis is characterized by degradation of ECM and restoration of normal liver histology. Following cessation of injury, there is a five-fold increase in collagenase activity in the liver, which is associated with a progressive decrease in the level of expression of TIMP-1 and TIMP-2, but no change in the level of expression of MMP-13. Studies in cultured HSCs have explored the molecular basis of the up-regulation of TIMP-1 during hepatic fibrogenesis. These studies have identified a key region in the TIMP-1 promoter that is critical to gene expression (32). However, up to now, it has not been identified the proteins interacting with this region. Many cell types, particularly HSCs, Kupffer cells and inflammatory cells, produce several MMPs in culture (20,26).
Very few studies have examined the effect of cytokines on ECM degradation in HSCs. However, it is well known that MMP production can be up-regulated by several hormones (PTH, cortisol), growth factors (platelet derived growth factor, basic fibroblast growth factor), cytokines (TNFa, IL-1a, IL-6, IL-10, IFNg, oncostatin M, macrophage migration inhibitory factor) and tumor promoters (phorbol esters). On the other hand, MMP-13 is down-regulated by transforming growth factor (TGFb), insulin-like growth factor (ILGF) (20,26,33-40). Likewise, several cytokines (TNFa, IL1b, IL-6, oncostatin M, IFNa) and growth factors (TGFb) up-regulate (41-43) or inhibit (44-47) TIMPs expression by cultured HSCs.
Interleukin-6 (IL-6) is a multifunctional glycoprotein produced by activated monocytes, macrophages, endothelial cells, and hepatic stellate cells that mediates a wide variety of functions in different cells, including fibroblasts, hepatocytes, and hepatic stellate cells (48). IL-6 promotes cell proliferation and differentiation and regulates specific gene expression (49), particularly the expression of the acute phase proteins in liver cells (48,50). Acute and chronic liver diseases, particularly alcoholic liver diseases, are similar to the acute phase response in some respects. Thus, patients with alcoholic hepatitis show fever, muscle wasting, neutrophilia, and increased production of C-reactive protein, a1-antitrypsin and amyloid A (50,51). High levels of IL-6 have been detected in the sera of patients with alcoholic liver cirrhosis (52-54) and some authors have shown a correlation between circulating concentrations of IL-6 and serum concentrations of C-reactive protein (55,56), an IL-6-induced protein. Thus, IL-6 seems to be one of the most important factors regulating inflammatory response in the liver.
We have shown that IL-6 increases MMP-13 protein levels and the steady-state levels of MMP-13 mRNA in a dose- and time-dependent fashion (Fig. 2) and that this effect is produced acting within the gene promoter (57). This effect is evident after 6 hours of treatment but it is particularly significant after 24 hours. This prolonged incubation time required by IL-6 to activate MMP-13 gene expression suggests that it may be required the synthesis of a new protein. The IL-6-induced increase in MMP-13 mRNA levels can be blocked by inhibiting the protein synthesis with cycloheximide (Fig. 2C), which supports this requirement.
This enhanced MMP-13 gene expression is associated with a marked increase in the immunoreactive MMP-13 protein (Fig. 2D). These results agree with those reported by Franchimont et al. (58) and Kusano et al. (59), who also demonstrated that IL-6, in the presence of its soluble receptor, increased MMP-13 mRNA levels, immunoreactive MMP-13 and its biological activity. It is likely that IL-6 has an effect on the extracellular metabolism of MMP-13, since the effect of this cytokine on MMP-13 secreted into the culture medium is much higher than that induced on the steady-state level of MMP-13 mRNA. Although some authors have shown that IL-6 significantly enhanced TIMP-1 production and TIMP-1 mRNA expression in human fibroblasts and other cell lines (60-64), in our experience, IL-6 also increases immunoreactive TIMP-1, but to a lesser extent (Fig. 2D). In fact, TIMP-1 and MMP-13 promoters contain common regulatory elements, i.e. an AP1 binding site (TRE) and PEA-3 (65). Therefore, factors that up-regulate MMP-13 gene expression may also stimulate TIMP-1 expression.
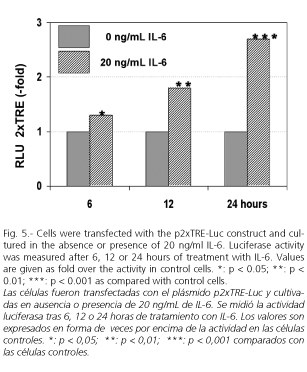
Rat MMP-13 promoter displays a general organization similar to that of other members of the MMP family (66,67), particularly to human (68) and rabbit MMP-1 genes (69). All share a common 10-exon organization (67,68,70) and contain a typical TATA box in addition to TRE and polyomavirus enhancer activator 3 (PEA-3) consensus site in their promoter region (67,71-74), suggesting a common regulatory mechanism of gene transcription. Rat MMP-13 gene also contains several consensus transcription factor recognition sequences such as C/EBP, Cbfa1, p53, AP-2, and AP1 (75) (Fig. 3). In cells transfected with plasmids whose activity is driven by portions of different length of the MMP-13 gene promoter, we found that sequences upstream of the base pair -79 are dispensable in IL-6-medited stimulation of MMP-13 gene expression. These experiments also show that integrity of the TRE site appears to be essential for the IL-6 induced response (Fig. 4). The TRE site has been implicated in the expression of many of the MMP genes (76,77), including MMP-13 (77). In cells transfected with a luciferase construct containing two copies of the TRE upstream of a minimal promoter, we were able to confirm this stimulatory effect of IL-6 on the TRE site (Fig. 5). Very little information exists about the effect of IL-6 on the activation of genes with a TRE. However, while Daffada et al. (78) found that IL-6 had no effect on the expression of pTRE-CAT in transient transfected cells, suggesting that AP1 is not induced by IL-6 treatment, Melamed et al. (79) showed that IL-6 induced the formation of a TRE-binding complex, which was abrogated by anti-Jun specific antibodies.


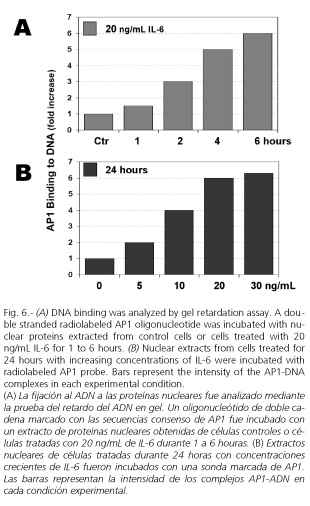
Upregulation of gene expression is usually induced by the binding of transcrition factors to specific cis-acting elements in gene promoters. Our study suggested that IL-6 upregulates MMP-13 gene expression via transcription factor AP1 and through the cis-acting element TRE. Thus, gel DNA retardation experiments show that IL-6 promotes a dose- and time-dependent binding of nuclear proteins to TRE site (Fig. 6). Nuclear protein-DNA complexes induced by the IL-6 treatment contain phosphorylated c-Jun as shown by the incubation of nuclear extract with a phosphorylated c-Jun-specific antibody prior to the gel retardation assay, which lead to the formation of two supershifted complexes. These results support the role played by TRE and AP1 in mediating the effect of IL-6 on rat MMP-13 gene expression.
The AP1 transcription factor is organized into Jun-Jun, Jun-Fos, or Jun-ATF dimmers, and the presence of Jun family members in these dimmers enables AP1 to bind cis-acting elements of gene promoters. These proteins form a variety of homo- and heterodimers that bind to a common DNA recognition site, the TRE (58,80,81). As figures 7 shows, culture of fibroblasts with IL-6 induces c-Jun, JunB and c-Fos mRNA (Fig. 7A), c-jun and c-fos promoters (Figs. 7B and 7C) and c-Jun and c-Fos proteins (Figs. 8A and 8B). Other authors have also shown that IL-6 stimulated junB gene expression in a variety of cells (82-86) and Cressman et al. (87) have shown that the expression of junB is markedly reduced in the liver of IL-6-deficient mice.
Transcriptional activity of AP1 depends not only on the quantity of AP1 components and their ability to bind DNA but also on the degree of phosphorylation of these proteins (80,88). Phosphorylation of c-Jun in its activation domain at serine 63 and 73 prolongs its half-life and potentiates the ability of c-Jun to activate transcription as either a homodimer or as a heterodimer with c-Fos (80). Western blot using a specific monoclonal antibody for serine 63 phosphorylated c-Jun demonstrates that IL-6 induces an increase in this form of c-Jun, which is particularly marked after 12 hours of treatment (Fig. 8C). Lütticken et al. (84) also showed that IL-6 triggers a delayed phosphorylation of STAT3 at serine residues. A variety of protein kinases, including JNK (c-Jun N-terminal kinase), pp42, pp54, pp44-, and p38-mitogen-activated protein kinases (89), p34cdc2, protein kinase C, casein kinase II, efficiently phosphorylates c-Jun (90). JNK, also known as stress-activated protein kinase (SAPK), is the most efficient member of the mitogen-activated protein kinase (MAPK) family involved in c-Jun phosphorylation on serines 63 and 73 of c-Jun and in potentiating its transactivation function (91,92). Moreover, JNK is a critical MAPK pathway for IL-1 induced collagenase gene expression in a variety of cell types (93). However, our study indicates that IL-6 does not induce the phosphorylation of c-Jun by stimulating JNK activity. As figure 9A shows incubation of cells with 10 and 20 ng/ml IL-6 for 15 minutes, but not for 5 minutes, decreased JNK activity to 84 and 68%, respectively, of that of control cells. Moreover, Western blots using specific polyclonal antibodies against JNK reveal one single band located at 46 kDa, the density of which does not change after incubation with IL-6. Likewise, this treatment do not modify phosphorylated JNK, as reveal Western blots using specific monoclonal IgG antibody raised against phosphorylated JNK (Fig. 9B). As dual phosphorylation of serine/threonine amino residues by the MAP kinase kinase (MKK4 and MKK7) is necessary for JNK activation (94), these results support the conclusion that this kinase is not involved in the IL-6-induced increase in phosphorylated c-Jun and MMP-13 gene expression. These results concurs with those reported by a number of authors showing that IL-6 has no detectable effects on JNK (95,96) or even inhibits the activation of this MAP kinase (97,98).
As mentioned above, in addition to JNK, a variety of protein kinases, including ERK, p38 kinase, casein kinase II, and PKC (99) can efficiently phosphorylate c-Jun (90,100). Moreover, in some cell lines, ERK pathway seems to play a role in mediating some effects of IL-6 (98). However, blocking these kinases with specific inhibitors does not abrogate the stimulatory effect of IL-6 on c-Jun phosphorylation (Fig. 10A), AP1 binding to DNA (Fig. 10B) or MMP-13 gene expression (Fig. 10C). In a recent study (101), we blocked ERK with PD98059, p38 MAP kinase with SB203580, protein kinase C (PKC) with H7 and GF 109203X, protein kinase A with H8 and H89, calmoduline-dependent protein kinase with calmidazolium, and phosphatidylinositol-3-kinase with wortmannin. Taken together, these results indicate that none of these protein kinases is likely required for the effect of IL-6 on MMP-13 gene expression.
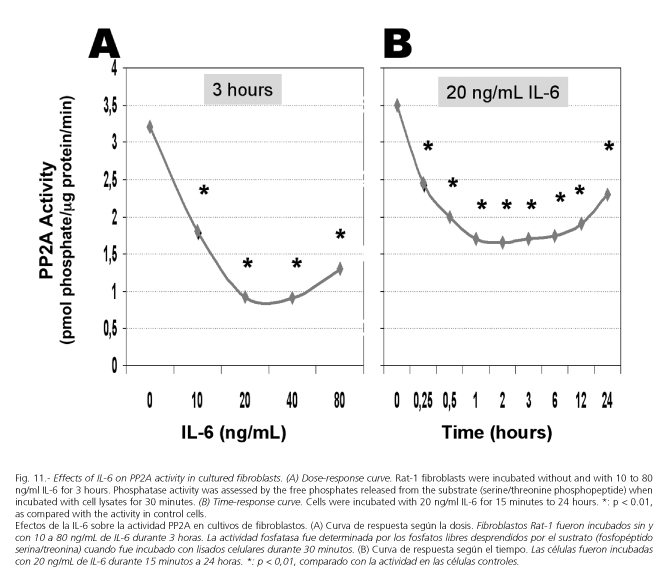
Phosphorylation state of c-Jun is a dynamic process controlled by both serine/threonine protein kinases and protein phosphatases 2A (PP2A) (102). Therefore, the increase in phosphorylated c-Jun may result not only by an enhanced kinase activity but also by a reduced PP2A activity. In fact, a number of studies have clearly demonstrated that inhibition of protein phosphatases 2A results in an induction of collagenase, JunB, and c-Fos mRNA and a potent activation of AP1, through serine/threonine phosphorylation (103-106). There are four classes of serine/threonine-specific protein phosphatases. These include PP1, PP2A, PP2B (calcineurin), and PP2C. PP2A and PP1 are widely distributed in the cytoplasm of mammalian cells and have been reported to be involved in signalling pathways, modifying the activity of a variety of protein kinases (107). Measurement of the PP2A activity in fibroblasts treated with increasing concentrations of IL-6 shows that this cytokine decreased this activity in a dose dependent fashion (Fig. 11A). Likewise, incubation of cells with 20 ng/ml IL-6 for 15 minutes to 24 hours resulted in a decrease in serine/threonine phosphatase activity, which was particularly marked between three and six hours. At 12 and 24 hours, this activity remained under the control activity (Fig. 11B).
The key role of PP2A in the IL-6-induced MMP-13 gene expression is also supported by experiments in which Rat-1 fibroblasts are pretreated for 20 minutes with 25 nM okadaic acid, a potent inhibitor of protein phosphatase 1 and PP2A (108). In this experimental condition, MMP-13-mRNA levels increase markedly and the addition of 20 ng/ml IL-6 for 24 hours does not modify this effect (Fig. 12). In contrast, as expected, inhibition of tyrosine phosphatases with 0.1 mM pervanadate does not change the stimulatory effects of IL-6 on MMP-13 gene expression. Furthermore, okadaic acid mimics the effects of IL-6 on MMP-13 gene expression. Many other authors have shown these effects of the okadaic acid on MMP gene expression, as well as on c-Jun phosphorylation and AP1 binding activity (105,109,110). I2PP2A, another potent inhibitor of PP2A, have produced similar effects (106). This decrease in PP2A activity is not due to a reduced amount of PP2A protein in fibroblasts exposed to IL-6. Thus, measurement of these levels in cell extracts from fibroblasts treated with 20 ng/ml IL-6 for three to sixteen hours demonstrates that this cytokine did not induce any change in intracellular PP2A levels.
Core structure of PP2A consists of complexes of three subunits (102,111,112). A 36 kDa catalytic C subunit (PP2AC) is complexed with a structural A subunit of 65 kDa (PP2A-A). This dimer is associated with a third, variable regulatory B subunit, that likely influences substrate specificity or cellular localization (102). The phosphorylation on tyrosine-307 at the C-terminus of the catalytic subunit of PP2A regulates phosphatases activity (113,114). It is well known that this phosphorylation is associated with a 90% loss in activity, and thereby with a sustained effect of protein kinases (113,115), including c-Jun phosphorylation. On the contrary, dephosphorylation of PP2AC reactivates this enzyme (102). Considering this mechanism of regulation of the PP2A activity, we could hypothesize that binding of IL-6 to specific receptors on cell surface might inactivate PP2A by increasing tyrosine phosphorylation on its catalytic subunit which, in turn, would prolong activation of AP1. Similar sequence of events has been shown following addition of insulin to skeletal muscle cells (115,116). Moreover, inhibition of PP2A with I2PP2A increased both the concentration and DNA binding of c-Jun and the transcriptional activity of AP1 (106). Likewise, PP2A inhibitors induce threonine phosphorylation of STAT3, other cytoplasmic transcription factor that upon activation translocate into the nucleus where they activate target genes (117,118).
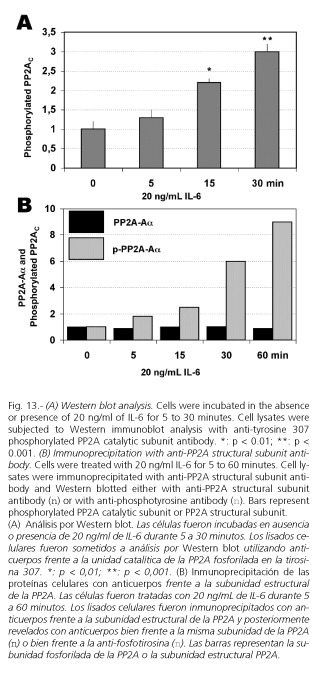
Western blot analysis using specific polyclonal antityrosine 307 phosphorylated PP2AC antibody shows a marked increase in the tyrosine phosphorylation of PP2AC (Fig. 13A). On the other hand, while immunoprecipitation of PP2A using specific antibody against the structural subunit of PP2A demonstrates that IL-6 did not affect to the total amount of this subunit, immunodetection of tyrosine phosphorylated proteins with antiphosphotyrosine antibody indicates that treatment of cells with 20 ng/mL IL-6 for 0 to 60 minutes induces a time-dependent increase in the PP2AC tyrosine phosphorylation (Fig. 13B). This result concurs with those reported by Choi et al. (119), who also found that IL-6 induced PP2A tyrosine phosphorylation.
IL-6 initiates its actions by binding to a specific receptor complex on the cell membrane. This complex is composed of two subunits: an 80-kDa binding protein and a 130-kDa transmembrane signal transducting component (gp130) (48). Other members of the IL-6 cytokine superfamily (IL-11, oncostatin M, leukemia inhibitory factor, ciliary neutrophic factor, cardiotropin-1, neutrophin-1/B.cell stimulating factor-3) (120-122) share the same gp130 receptor subunit. Activation of IL-6 signal transduction involved gp130-homodimerization and tyrosine phosphorylation of the cytoplasmic tail of gp130 by gp130-associated Janus tyrosine kinases (JAKs). These kinases are recruited to the plasma membrane and activated upon cell activation by cytokines and growth factors (123). All JAKs contain at least one copy of a peptide sequence, which is part of a consensus for PP2A binding (124,125). This phosphatase has been shown to form stable complexes with various protein kinases, including Jak2 (115,126,127). Thus, it is conceivable that the IL-6-induced PP2AC phosphorylation and PP2A inactivation might be mediated by Jak2. Thus, as figure 14 shows, pre-treatment of cells for 60 minutes with 5 µM AG490, a specific inhibitor of Jak2 (128), abrogates completely the IL-6-induced increase in MMP-13 mRNA, binding of nuclear proteins to AP1 consensus sequence, c-Jun and PP2AC phosphorylation, and PP2A activity. As far as we are aware, no further information is available on the role of Jak kinases and PP2A in mediating the effect of IL-6 on MMP-13 gene expression. However, oncostatin M, another member of IL-6 superfamily cytokine (122), also induces MMP gene expression in astrocytes, fibroblasts and osteoblasts (37,130), and this effect is associated with Jak1 and Jak2 tyrosine phosphorylation.
Thus, we can conclude that, at least "in vitro", IL-6 is an antifibrogenic cytokine that might contribute to remodelling connective tissue. IL-6 increases MMP-13 gene expression acting at transcriptional level via AP1. IL-6 increases c-jun and c-fos gene expression, enhances phosphorylated c-Jun, and promotes the binding of AP1 to the MMP-13 promoter. The increase in phosphorylated c-Jun is not due to increased protein kinase activity, including Jun N-terminal kinase, but to a decreased serine/threonine phosphatases activity. IL-6, after binding to a specific receptor on the cell membrane, activates tyrosine kinase JAK2, which phosphorylates the catalytic subunit of protein phosphatases 2A (PP2A) at the tyrosine 307 leading to inhibition of its activity and prolongs c-Jun phosphorylation (Fig. 15).
REFERENCES
1. Orrego H, Blendis LM, Crossley IR, Medline A, Macdonald A, Ritchie S, et al. Correlation of intrahepatic pressure with collagen in the Disse space and hepatomegaly in humans and in the rat. Gastroenterology 1981; 80: 546-56. [ Links ]
2. Horn T, Christoffersen P, Henriksen JH. Alcoholic liver injury: defenestration in noncirrhotic liver- a scanning electron microscopic study. Hepatology 1987; 7: 77-82. [ Links ]
3. Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liv Dis 2001; 351-72. [ Links ]
4. Rojkind M, Greenwel P. The liver as a bioecological system. In: Arias IM, Jakoby WB, Popper H, Schachter D, Shafritz DA, eds. "The liver: Biology and Pathology". 2nd ed. New York: Raven Pres Ltd. 1988. p. 707-16. [ Links ]
5. Rojkind M, Giambrone MA, Biempica L. Collagen type in normal and cirrhotic liver. Gastroenterology 1970; 76: 710-9. [ Links ]
6. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000; 275: 2247-50. [ Links ]
7. Blomhoff R, Wake K. Perisinusoidal stellate cells of the liver: important roles in retinal metabolism and fibrosis. FASEB J 1991; 5: 271-7. [ Links ]
8. Kingsley DM, The TGFb superfamily: a new members, new receptors, and new genetic tests of function in different organisms. Genes Dev 1994; 8: 133-46. [ Links ]
9. Friedman SL. Cytokines and fibrogenesis. Semin Liv Dises 1999; 129-40. [ Links ]
10. Geerts A, Vrijsen R, Rauterberg J, Burt A, Schellinck P, Wisse E. In vitro transition of fat-storing cells to myofibroblast-like cells parallels marked increase of collagen synthesis and secretion. J Hepatol 1989; 9: 59-68. [ Links ]
11. Abdel-Aziz G, Lebeau G, Rescan PY, Clement B, Rissel M, Deugnier Y, et al. Reversibility of hepatic fibrosis in experimentally induced cholestasis in rat. Am J Pathol 1990; 137: 1333-43. [ Links ]
12. Morcos SH, Khayyal MT, Mansour MM, Saleh S, Ishak EA, Girgis NI, et al. Reversal of hepatic fibrosis after praziquantel therapy of murine schistosomiasis. Am Soc Trop Med Hygiene 1985; 34: 314-21. [ Links ]
13. Schiff ER, Heathcote J, Dienstag JL, et al. Improvements in liver histology and cirrhosis with extended lamivudine therapy. Hepatology 2000; 32: 296 (Abstract). [ Links ]
14. Hammel P, Couvelard A, O'Toole D, Ratouis A, Sauvanet A, Flejou JF, et al. Regression of liver fibrosis after biliary drainage in patients with chronic pancreatitis and stenosis of the common bile duct. N Engl J Med 2001; 344: 418-23. [ Links ]
15. Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med 2000; 124: 1599-607. [ Links ]
16. Iredale JP. Hepatic stellate cell behaviour during resolution of liver injury. Semin Liver Dis 2001; 21: 427-36. [ Links ]
17. Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med 2000; 124: 1599-607. [ Links ]
18. Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MDA, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology 2004; 126: 1795-808. [ Links ]
19. Zavalova L. Enzymes from the medicinal leech (Hirudo medicinalis) coding for the unusual enzymes that specifically cleave endoepsi (gamma-glut) lys isopeptide bonds and help to dissolve blood clots. Mol Cell Genet 1996; 253: 20-5. [ Links ]
20. Arthur MJ. Collagenases and liver fibrosis. J Hepatol 1995; 22: 44-8. [ Links ]
21. Iimuro Y, Nishio T, Morimoto T, Nitta T, Stefanovic B, Choi SK, et al. Delivery of matrix metalloproteinase-1 attenuates established liver fibrosis in the rat. Gastroenterology 2003; 124: 445-58. [ Links ]
22. Emonard H, Grimaud JA. Matrix metalloproteinases. Cell Mol Biol 1990; 36: 131-53. [ Links ]
23. Mengshol JA, Mix KS, Brinckerhoff CE. Matrix metalloproteinases as therapeutic targets in arthritis diseases. Arthritis Rheum 2002; 46: 13-20. [ Links ]
24. Nagase H, Woessner JF Jr. Matrix metalloproteinases. J Biol Chem 1999; 274: 21491-4. [ Links ]
25. Knäuper V, Cowell S, Smith B, López-Otín C, O'Shea M, Morris H, et al. The role of the terminal domain of human collagenase-3 (MMP-13) in the activation of procollagenase-3, substrate specificity, and tissue inhibitor of metalloproteinase interaction. J Biol Chem 1997; 272: 7608-16. [ Links ]
26. Mauviel A. Cytokine regulation of metalloproteinase gene expression. J Cell Biochem 1993; 53: 288-95. [ Links ]
27. Arthur MJP, Iredale JP, Mann DA. Inhibitors of metalloproteinases: role in liver fibrosis and alcoholic liver disease. Alcohol Clin Exp Res 1999; 23: 940-3. [ Links ]
28. Howard EW, Banda MJ. Binding of tissue inhibitor of metalloproteinases 2 to two distinct sites on human 72-kDa gelatinase. J Biol Chem 1991; 266: 17972-7. [ Links ]
29. Murphy FR, Issa R, Zhou X, Ratnarajah S, Nagase H, Arthur MJ, et al. Inhibition of apoptosis od activated hepatic stellate cells by tissue inhibitor of metalloproteinase-I is mediated via effects on matrix metalloproteinase inhibition. Implications for reversibility of liver fibrosis. J Biol Chem 2002; 277: 11069-76. [ Links ]
30. Murphy F, Waung J, Collins J, Arthur MJ, Nagase H, Mann D, et al. N-Cadherin cleavage during activated hepatic stellate cell apoptosis is inhibited by tissue inhibitor of metalloproteinase-1. Comp Hepatol 2004; 3 (Supl. 1): S8. [ Links ]
31. Knittel T, Mehde M, Grundmann A, Saile B, Scharf JG, Ramadori G. Expression of matrix metalloproteinases and their inhibitors during hepatic tissue repair in the rat. Histochem Cell Biol 2000; 113: 443-53. [ Links ]
32. Trim JE, Samra SK, Arthur MJ, Wright MC, McAulay M, Beri R, et al. Upstream tissue inhibitor of metalloproteinases-1 (TIMP-1) element-1, a novel and essential regulatory DNA motif in the human TIMP-1 gene promoter, directly interacts with a 30-kDa nuclear protein. J Biol Chem 2000; 275: 6657-63. [ Links ]
33. Siwik DA, Chang DL-F, Colucci WS. Interleukin-1 β and tumor necrosis factor-α decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Cir Res 2000; 86: 1259-65. [ Links ]
34. Wang SC, Ohata M, Schrum L, Rippe RA, Tsukamoto H. Expression of interleukin-10 by in vitro and in vivo activated hepatic stellated cells. J Biol Chem 1998; 273: 302-8. [ Links ]
35. Brenner DA, O´Hara M, Angel P, Chojkier M, Karin M. Prolonged activation of jun and collagenase genes by tumor necrosis factor-α . Nature 1989; 337: 661-3. [ Links ]
36. Delany AM, Jeffrey JJ, Canalis E. Cortisol increases interstitial collagenase expression in osteoblasts by post-transcriptional mechanisms. J Biol Chem 1995; 270: 26607-12. [ Links ]
37. Varghese S, Yu K, Canalis E. Leukemia inhibitory factor and oncostatin M stimulate collagenase-3 expression in osteoblasts. Am J Physiol 1999; 276: 465-71. [ Links ]
38. Onodera S, Nishihira J, Iwabuchi K, Koyama Y, Yoshida K, Tanaka S, et al. Macrophage migration inhibitory factor up-regulates matrix metalloproteinase-9 and -13 in rat osteobasts. J Biol Chem 2002; 277: 7865-74. [ Links ]
39. Gazzero E, Rydziel S, Canalis E. Skeletal bone morphogenetic proteins suppress the expression of collagenase-3 by rat osteoblasts. Endocrinology 1999; 140: 562-7. [ Links ]
40. Van den Berg WB. Pathophysiology of osteoarthritis. Joint Bone Spine 2000; 67: 555-6. [ Links ]
41. Knittel T, Mehde M, Kobold D, Saile B, Dinter C, Ramadori G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and nonparenchymal cells of rat liver: regulation by TNF-α and TGF-β 1. J Hepatol 1999; 30: 48-60. [ Links ]
42. Herbst H, Wege T, Milani S, Pellegrini G, Orzeckowski HD, Bechstein WO, et al. Tissue inhibitor of metalloproteinase-1 and -2 RNA expression in rat and human liver fibrosis. Am J Pathol 1997; 150: 1647-59. [ Links ]
43. Rieder H, Armbrust T, Meyer zum Buschenfelde KH, Ramadori G. Contribution of sinusoidal endothelial liver cells to liver fibrosis: expression of transforming growth factor-beta 1 receptors and modulation of plasmin-generating enzymes by transforming growth factor-beta 1. Hepatology 1993; 18: 937-44. [ Links ]
44. Sakaida I, Uchida K, Matsumura Y, Okita K. Interferon gamma treatment prevents procollagen gene expression without affecting transforming growth factor-b1 expression in pig serum-induced rat liver fibrosis in vivo. J Hepatol 1998; 28: 471-9. [ Links ]
45. Li YY, McTiernan CF, Feldman AM. Proinflammatory cytokines regulate tissue inhibitors of metalloproteinases and disintegrin metal-loproteinase in cardiac cells. Cardiovasc Res 1999; 42: 162-72. [ Links ]
46. Arai M, Niioka M, Maruyama K, Wada N, Fujimoto N, Nomiyama T, et al. Changes in serum levels of metalloproteinases and their inhibitors by treatment of chronic hepatitis C with interferon. Dig Dis Sci 1996; 41: 995-1000. [ Links ]
47. Mitsuda A, Suou T, Ikuta Y, Kawasaki H. Changes in serum tissue inhibitor of matrix metalloproteinase-1 after interferon alpha treatment in chronic hepatitis C. J Hepatol 2000; 32: 666-72. [ Links ]
48. Hirano T. Interleukin-6 and its receptor: ten year later. Int Rev Immunol 1998; 16: 249-84. [ Links ]
49. Taga T, Kishimoto T. Human cytokines. In: Aggarwal BB, Gutterman JU, eds. Handbook for basic and clinical research. Oxford, United Kingdom: Blackwell Scientific Publications, 1992. p. 144-63. [ Links ]
50. Thiele DL. Tumor necrosis factor, the acute phase response and the pathogenesis of alcoholic liver disease. Hepatology 1989; 9: 497-9. [ Links ]
51. Sweeting J. Tumor necrosis factor in alcoholic hepatitis. Gastroenterology 1989; 97: 1056-7. [ Links ]
52. Deviere J, Content J, Denys C, Vandenbussche P, Le Moine O, Schandene L, et al. Immunoglobulin A and interleukin 6 form a positive secretory feedback loop: a study of normal subjects and alcoholic cirrhotics. Gastroenterology 1992; 103: 1296-301. [ Links ]
53. Díez-Ruiz A, Santos-Pérez JL, López-Martínez G, González-Calvín J, Gil-Estremera B, Gutiérrez-Gea F. Tumor necrosis factor, interleukin-1 and interleukin-6 in alcoholic cirrhosis. Alcohol Alcohol 1993; 28: 319-23. [ Links ]
54. Khoruts A, Stahnke L, McClain CJ, Logan G, Allen JI. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology 1991; 13: 267-76. [ Links ]
55. Barber MD, Fearon KC, Ross JA. Relationship of serum levels of interleukin-6, soluble interleukin-6 receptor and tumor necrosis factor receptors to the acute-phase protein response in advanced pancreatic cancer. Clin Sci (Lond) 1999; 96: 83-7. [ Links ]
56. Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, Van Snick J. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum 1988; 31: 784-7. [ Links ]
57. Solis-Herruzo JA, Rippe RA, Schrum LW, De la Torre P, García I, Jeffrey JJ, et al. Interleukin-6 rat metalloproteinase-13 gene expression through stimulation of activator protein 1 transcription factor in cultured fibroblasts. J Biol Chem 1999; 274: 30919-26. [ Links ]
58. Franchimont N, Rydziel S, Delany AM, Canalis E. Interleukin-6 and its soluble receptor cause a marked induction od collagenase 3 expression in rat osteoblest cultures. J. Biol. Chem 1997; 272: 12144-50. [ Links ]
59. Kusano K, Miyaura C, Inada M, Tamura T, Ito A, Nagase H, et al. Regulation of matriz metalloproteinases (MMP-2, -3, -9, and -13) by interleukin-1 and interleukin-6 in mouse calvaria: association of MMP induction with bone resorption. Endocrinology 1998; 139: 1338-45. [ Links ]
60. Shingu M, Nagai Y, Isayama T, Naono T, Nobunaga M, Nagai Y. The effects of cytokines on metalloproteinase inhibitors (TIMP) and collagenase production by human chondrocytes and TIMP production by synovial cells and endothelial cells. Clin Exp Immunol 1993; 94: 145-9. [ Links ]
61. Lotz M, Guerne PA. Interleukin-6 induces the synthesis of tissue inhibitor of metalloproteinases-1/erythroid potentiating activity (TIMP-1/ EPA). J Biol Chem 1991; 266: 2017-20. [ Links ]
62. Sato T, Ito A, Mori Y. Interleukin-6 enhances the production of tis-sue inhibitor of metalloproteinases (TIMP) but not that of matrix metalloproteinases by human fibroblasts. Biochem Biophys Res Commun 1990; 170: 824-9. [ Links ]
63. Silacci P, Dayer JM, Desgeorges A, Peter R, Manueddu C, Guerne PA. Interleukin (IL)-6 and its soluble receptor induce TIMP-1 expression in synoviocytes and chondrocytes, and block IL-1-induced collagenolytic activity. J Biol Chem 1998; 273: 13625-9. [ Links ]
64. Richards CD, Shoyab M, Brown TJ, Gauldie J. Selective regulation of metalloproteinase inhibitor (TIMP-1) by oncostatin M in fibroblasts in culture. J Immunol 1993; 150: 5596-608. [ Links ]
65. Edwards DR, Rocheleau H, Sharma RR, Wills AJ, Cowie A, Hassel JA, et al. Involment of AP1 and PEA3 binding site in the regulation of murine tissue inhibitor of metalloproteinases-1 (TIMP-1) transcription. Biochim Biophys Acta 1992; 1171: 41-55. [ Links ]
66. Matrisian LM, Glaichenhaus N, Gesnel MC, Breathnach R. Epidermal growth factor and oncogenes induce transcription of the same cellular mRNA in rat fibroblasts. EMBO J 1985; 4: 1435-40. [ Links ]
67. Pendás AM, Balbín M, Llano E, Jiménez MG, López-Otín C. Structural analysis and promoter charactarization of the human collagenase-3 gene (MMP-13). Genomics 1997; 40: 222-33. [ Links ]
68. Collier IE, Smith J, Kronberger A, Bauer EA, Wilhelm SM, Eisen AZ, et al. The structure of the human skin fibroblast collagenase gene. J Biol Chem 1988; 263: 10711-3. [ Links ]
69. Fini ME, Plucinska IM, Mayer AS, Gross RH, Brinckerhoff CE. A genes for rabbit synovial cell collagenase: member of a family of metalloproteinases that degrade the connective tissue matrix. Biochemistry 1987; 26: 6156-65. [ Links ]
70. Belaaouaj A, Shipley JM, Kobayashi DK, Zimonjic DB, Popescu N, Silverman GA, et al. Human macrophage metalloelastase. Genomic organization, cjromosomal lacation, gene linkage, and tissue-specific expression. J Biol Chem 1995; 270: 14568-75. [ Links ]
71. Rajakumar RA, Quinn CO. Parathyroid hormone induction of rat interstitial collagenase mRNA in osteosarcoma cells is mediated through an AP-1-binding site. Mol Endo 1996; 10: 867-78. [ Links ]
72. Huhtala P, Tuuttila A, Chow LT, Lohi J, Keski-Oja J, Tryggvason K. Complete structure of the human gene for 92-kDa type IV collagenase. Divergent regulation of expression for the 92- and 72- kilodal-ton enzyme genes in HT-1080 cells. J Biol Chem 1991; 266: 16485-90. [ Links ]
73. Gaire M, Magbanua Z, McDonnell S, McNeil L, Lovett DH, Matrisian LM. Structure and expression of the human gene for the matrix metalloproteinase matrilysin. J Biol Chem 1994; 269: 2032-40. [ Links ]
74. Tardif G, Pelletier JP, Dupuis M, Hambor JE, Martel-Pelletier J. Cloning, sequencing and characterization of the 5-flaking region of the human collagenase-3 gene. Biochem J 1997; 323: 13-6. [ Links ]
75. Selvamurugan N, Chou WY, Pearman AT, Pulumati MR, Partridge NC. Parothyroid hormone regulates the rat collagenase-3 promoter in osteoblastic cells through the cooperative interaction of the activator protein-1 site and the runt domain binding sequence. J Biol Chem 1998; 273: 10647-57. [ Links ]
76. Angel P, Baumann I, Stein B, Delius H, Rahmsdorf HJ, Herrlich P. 12-O-tetradecanoyl-phorbol-13-acetate induction of the human colla-genase gene is mediated by an inducible enhancer element located in the 5-flaking region. Mol Cell Biol 1987; 7: 2256-66. [ Links ]
77. Borden P, Solymar D, Sucharczuk A, Lindman B, Cannon P, Heller RA. Cytokine control of interstitial collagenase and collagenase-3 gene expression in human chondrocytes. J Biol Chem 1996: 271: 23577-81. [ Links ]
78. Daffada AA, Murray EJ, Young SP. Control of activator protein-1 and nuclear factor kappa B activity by interleukin-1, interleukin-6 and metals in HEPG2 cells. Biochim Biophys Acta 1994; 1222: 234-40. [ Links ]
79. Melamed D, Resnitzky D, Haimov I, Levy N, Pfarr CM, Yaniv M, et al. Interleukin 6 induces DNA binding activity of AP1 in M1 myeloblastic cells but not in a growth resistant cell derivative. Cell Growth Differ 1993; 4: 689-97. [ Links ]
80. Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem 1995; 270: 16483-6. [ Links ]
81. Trenies I, Paterson HF, Hooper S, Wilson R, Marshall CJ. Activated MEK stimulates expression of AP1 components independently of phosphatidylinositol 3-kinase (PI3-kinase) but requires a PI3-kinase signal to stimulate DNA synthesis. Mol Cell Biol 1999; 19: 321-9. [ Links ]
82. Brown RT, Ades IZ, Nordan RP. An acute phase response factor/NF-ka`ppa B site downstream of the junB gene that mediates responsiveness to interleukin-6 in a murine plasmacytoma. J Biol Chem 1995; 270: 31129-35. [ Links ]
83. Wang Y, Fuller, GM. Interleukin-6 and ciliary neurotrophic factor trigger janus kinase activation and early gene response in rat hepatocytes. Gene 1995; 162: 285-9. [ Links ]
84. Lütticken C, Coffer P, Yuan J, Schwartz C, Caldenhover E, Schindler C, et al. Interleukin-6-induced serine phosphorylation of transcription factor APRF: evidence for a role in interleukin-6 target gene induction. FEBS letters 1995; 360: 137-43. [ Links ]
85. Jenab S, Morris PL. Transcriptional regulation of Sertoli cell immediate early genes by interleukin-6 and interferon-gamma is mediated through phosphorylation of STAT-3 and STAT-1 proteins. Endocrinology 1997; 138: 2740-6. [ Links ]
86. Kojima H, Nakajima K, Hirano T. IL-6-inducible complexes on an IL-6 response element of the junB promoter contain Stat3 and 36 kDa CRE-like site binding protein(s). Oncogene 1996; 12: 547-54. [ Links ]
87. Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice Science 1996; 274: 1379-83. [ Links ]
88. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001; 410: 37-40. [ Links ]
89. Ahmed S, Rahman A, Hasnain A, Goldberg VM, Haqqi TM. Phenyl N-tert-butylnitrone down-regulates interleukin-1 β -stimulated matrix metalloproteinase-13 gene expression in human chondrocytes: suppression of c-Jun NH2-terminal kinase, p38-mitogen-activated protein kinase and activating protein-1. J Pharmacol Exper Ther 2003; 305: 981-8. [ Links ]
90. Baker SJ, Kerppola TK, Luk D, Vandenberg MT, Marshak JR, Curran T et al. Jun is phosphorylated by several protein kinases at the same sites that are modified in serum-stimulated fibroblasts. Mol Cell Biol 1992; 12: 4694-705. [ Links ]
91. Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein-and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev 1993; 7: 2135-48. [ Links ]
92. Karin M. The regulation of AP1 activity by mitogen-activated protein kinase. J Biol Chem 1995; 270: 16483-6. [ Links ]
93. Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, et al. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest 2001; 108: 73-81. [ Links ]
94. Davis RJ. Signal transduction by the c-Jun terminal kinase. Biochem Soc Symp 1999; 64: 1-12. [ Links ]
95. D´Arcangelo G, Tancredi V, Onofri F, DAntuono M, Gioverdi S, Benfenati F. Interleukin-6 inhibits neurotransmitter release and the spread of excitation in the rat cerebral cortex. Eur J Neurosci 2000; 12: 1241-52 [ Links ]
96. Tancredi V, D´Antuono M, Cafe C, Giovedi S, Bue MC, D´Archangelo G, et al. The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem 2000; 75: 634-43. [ Links ]
97. Shioda S, Ozawa H, Dohi K, Mizushima H, Matsumoto K, Nakajo S, et al. PACAP protects hippocampal neurons against apoptosis: involvement of JNK/SAPK signaling pathway. Ann NY Acd Sci 1998; 865: 111-7. [ Links ]
98. Xu FH, Sharma S, Gardner A, Tu Y, Raitano A, Sawyers C, et al. Interleukin-6-induced inhibition of multiple myeloma cell apoptosis: support for the hypotesis that protection is mediated via inhibition of the JNK/SAPK pathway. Blood 1998; 92: 241-51. [ Links ]
99. Adler V, Franklin CC, Kraft AS. Phorbol esters stimulate the phosphorylation of c-Jun but not v-Jun: regulation by the N-terminal delta domain. Proc Natl Acad USA 1992; 89: 5341-5. [ Links ]
100. Ye J, Zeidler P, Young SH, Martínez A, Robinson VA, Jones W, et al. Activation of mitogen-activated protein kinase p38 and extracellular signal-regulated kinase is involved in glass fiber-induced tumor necrosis factor-alpha production in macrophages. J Biol Chem 2001; 276: 5360-7. [ Links ]
101. De la Torre P, Díaz-Sanjuán T, García-Ruiz I, Esteban E, Canga F, Muñoz-Yagüe T, et al. Interleukin-6 increases rat metalloproteinase-13 gene expression through Janus kinase-2-mediated inhibition of serine/threonine phosphatase-2A. Cellular Signalling 2005; 17: 427-35. [ Links ]
102. Wera S, Hemmings BA. Serine/threonine protein phosphatases. Biochem J 1995; 311: 17-29. [ Links ]
103. Westermarck J, Lohi J, Keski-Oja J, Kaharri VM. Okadaic acidelicited transcriptional activation of collagenase gene expression in HT-1080 fibrosarcoma cells is mediated by JunB. Cell Growth Differ 1994; 5: 1205-13. [ Links ]
104. Kim SJ, Lafyatis R, Kim KY, Angel P, Fujiki H, Karim M, et al. Regulation of collagenase gene expression by okadaic acid, an inhibitor of protein phosphatases. Cell Regul 1990; 1: 269-78. [ Links ]
105. Thévenin C, Kim S-J, Kehrl JH. Inhibition of protein phosphatases by okadaic acid induces AP1 in human T cells. J Biol Chem 1991; 266: 9363-6. [ Links ]
106. Al-Murrani SW, Woodgett JR, Damuni Z. Expression of I2PP2A, an inhibitor of protein phosphatases 2A, induces c-Jun and AP-1 activity. Biochem J 1999; 341: 293-8. [ Links ]
107. Mumby MC, Walter G. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol Rev 1993; 73: 673-99. [ Links ]
108. Cohen P, Holmes CFB, Tsukitani Y. Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem Sci 1990; 15: 98-102. [ Links ]
109. Peng J, Bowden GT, Domann FE. Activation of AP-1 by okadaic acid in mouse keratinocytes associated with hyperphosphorylation of c-jun. Mol Carcinog 1997; 18: 37-43. [ Links ]
110. Grumbles RM, Shao L, Jeffrey JJ, Howell DS. Regulation of rat interstitial collagenase gene expression in growth cartilage and chondrocytes by vitamin D3, interleukin-1 beta, and okadaic acid. J Cell Biochem 1996; 63: 395-409. [ Links ]
111. Csortos C, Zolnierowicz S, Bako E, Durbin SD, DePaoli-Roach AA. High complexity in the expression of the B subunit of protein phosphatases 2A. Evidence for the existence of at least seven novel iso-forms. J Biol Chem 1996; 271: 2578-88 [ Links ]
112. Zolnierowicz S. Type 2A protein phosphatase, the complex regulator of numerous signaling pathways. Bichem Pharmacol 2000; 60: 1225-35. [ Links ]
113. Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatases type-2A by tyrosine phosphorylation. Science 1992; 257: 1261-4. [ Links ]
114. Chen J, Parsons S, Brautigan DL. Tyrosine phosphorylation of protein phosphatases 2A in response to growth stimulation and v-src transformation of fibroblasts. J Biol Chem 1994; 269: 7957-62. [ Links ]
115. Begum N, Ragolia L. Role of janus kinase-2 in insulin-mediated phosphorylation and inactivation of protein phosphatase-2A and its impact on upstream insulin signalling components. Biochem J 1999; 344: 895-901. [ Links ]
116. Srinivasan M, Begum N. Stimulation of protein phosphatase –1 acttivity by phorbol esters. Evaluation of the regulatory role of protein kinase C in insulin action. J Biol Chem 1994; 269: 16662-7. [ Links ]
117. Liu KD, Gaffen SL, Goldsmith MA. JAK/STAT signaling by cytokine receptors. Curr Immunol 1998; 10: 271-8. [ Links ]
118. Woetmann A, Nielsen M, Christiansen ST, Brockdorff J, Kaltoft K, Engel AM, et al. Inhibition of protein phosphatase 2A induces serine/threonine phosphorylation, subcellular redistribution, and functional inhibition of STAT3. Proc Natl Acad Sci USA 1999; 96: 10620-5. [ Links ]
119. Choi I, Lee MJ, Kim EJ, Kang HS, Pyun KH. Roles of protein phosphatase 2A in IL-6 signal transduction in Hep3B cells. Immunol Lett 1998; 61: 103-7. [ Links ]
120. Gómez-Lechón MJ. Oncostatin M: signal transduction and biological activity. Life Sci 1999; 65: 2019-30. [ Links ]
121. Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol 1997; 15: 797-819. [ Links ]
122. Miyajima A, Kinoshita T, Tanaka T, Kamiya A, Mukuoyama Y, Hara T. Role of Oncostatin M in hematopoiesis and liver develop-ment. Cytokine Growth Factor Rev 200; 11: 177-83. [ Links ]
123. Saad MJ, Carlvaho CR, Thirone AC, Velloso LA. Insulin induces tyrosine phosphorylation of JAK2 in insulin-sensitive tissues of the intact rat. J Biol Chem 1996; 271: 22100-4. [ Links ]
124. Glenn G, Eckhart W. Amino-terminal regions of polyomavirus middle T antigen are required for interactions with protein phosphatase 2A. J Virol 1995; 69: 3792-36. [ Links ]
125. Yokoyama N, Reich NC, Miller WT. Determinants for the interaction between Janus kinase 2 and protein phosphatase 2A. Arch Biochem Biophys 2003; 417: 87-95. [ Links ]
126. Yokoyama N, Reich NC, Miller WT. Involvement of protein phosphatase 2A in the interleukin-3-stimulated Jak2-Stat5 signaling path-way. J Interferon Cytokine Res 2001; 21: 369-78. [ Links ]
127. Fuhrer DK, Yang YC. Complex formation of JAK2 with PP2A, P13K, and Yes in response to the hematopoietic cytokine interleukin-11. Biochem Biophys Res Commum 1996; 224: 289-96. [ Links ]
128. Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 1996; 379: 645-8. [ Links ]

