










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.98 n.9 Madrid Sep. 2006
CLINICAL NOTE
Congenital hepatic fibrosis associated with von Recklinghausen's disease
Fibrosis hepática congénita asociada a enfermedad de von Recklinghausen
O. A. Jorge and A. D. Jorge
Service of Gastroenterology. Hospital Español. Mendoza. Argentina
ABSTRACT
Congenital hepatic fibrosis is characterized by a ductal plate malformation with duct-like structures and fibrosis. It manifests clinically with portal hypertension and may be associated with multiple congenital defects. We present the case of a 16-year-old male with splenomegaly, leukopenia and thrombocytopenia, esophageal varices, and a histopathological diagnosis of congenital hepatic fibrosis. He exhibits "café au lait' spots and "Lisch' nodules, with a diagnosis of von Recklinghausen's disease. Congenital hepatic fibrosis belongs to the so-called fibropolycystic diseases, in which there is a disordered interaction between cells and the extracellular matrix. Von Recklinghausen's disease affects tissues derived from the neural crest and its diagnosis is based on clinical criteria. It is associated with multiple diseases. We describe its association with congenital hepatic fibrosis for the first time.
Key words: Congenital hepatic fibrosis. Portal hypertension. Von Recklinghausen's disease.
Introduction
Congenital hepatic fibrosis (CHF) is an autosomal recessive disease that may be familial or sporadic (1).
In CHF there is a malformation of the ductal plate, which is a circular embryonic structure appearing in the eighth week of gestation that is formed by primitive hepatocytes, which differentiate into cholangiocytes. The ductal plate surrounds the mesenchyme of portal tracts and, after a process of extensive involution and remodelling, intrahepatic bile ducts develop (2). As a consequence of this malformation, with persistence of abnormal portions and remodelling areas in the ductal plate, there are ductal-like structures of biliary origin and fibrosis that do not alter hepatic architecture. This fibrosis would affect venous resistance in portal branches, thus developing portal hypertension. There would also be abnormalities in intrahepatic portal branches with a reduction in size (3).
CHF clinically manifests during childhood and adolescence with hepatomegaly, splenomegaly, and signs of portal hypertension such as esophageal varices. It may be associated with congenital defects of the kidney (cystic dilatations of the distal tubules and collecting ducts), cerebellum (hemangiomas), lung (emphysema), heart, vessels (aneurysms), or intestines (lymphangiectasia) (4).
We present its association with von Recklinghausen's disease for the first time.
Case report
A 16-year-old boy with no personal or familial history of disease consulted for thrombocytopenia and a diagnosis of hepatic cirrhosis. At physical examination we saw multiple "café au lait' spots all over his trunk (1-7 cm in size) (Fig. 1) and marked splenomegaly. Laboratory findings: leukocytes 3,700/mm3; platelets 52,000/mm3; creatinine 0.76 mg/dl; uremia 26 mg/dl; prothrombin time 100% (n.v. 70-110); AST 30 U/l (n.v. up to 40); ALT 48 U/l (n.v. up to 40); AP 174 U/l (n.v. up to 306); total bilirubin 1.20 mg/dl (n.v. up to 1); albumin 4.17 g/dl (n.v. 3.5-5); HBsAg (-); anti-HBc (-); Anti-HCV (-); ANA (-); AMA (-); ASMA (-); ceruloplasmin, alpha 1-antitrypsin, percentage transferrin saturation, and total bile acids within normal values. Ultrasonography: liver with multiple echogenic linear images (compatible with fibrosis) that circumscribe pseudonodular areas. Dilated portal vein (15 mm) and umbilical vein rechanneled with hepatofugal blood flow. Intrahepatic veins in the portal venous system were small and with a low flow. The gallbladder and bile ducts were normal. Marked homogeneous splenomegaly (19 cm). Kidneys were normal. No ascites.
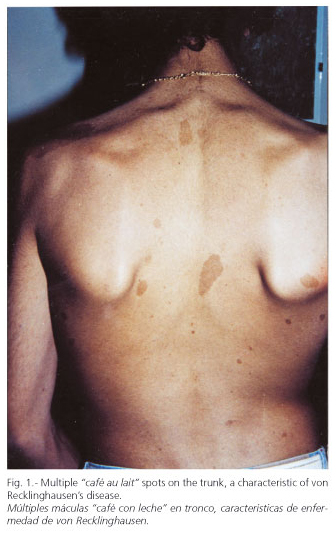
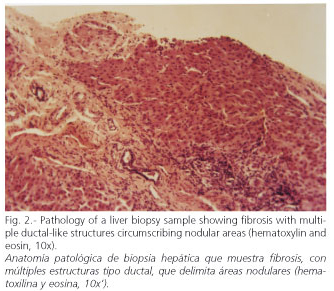
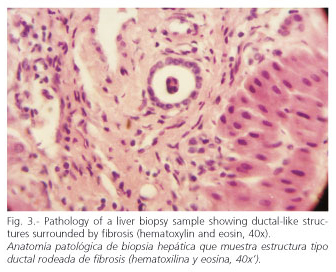
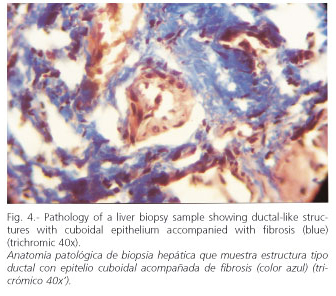
Endoscopy: grade-I esophageal varices. Laparoscopy: liver with a nodular aspect and fibrous bands. Hepatic biopsy: areas of periportal fibrosis, with ductal-like structures of cuboidal epithelium, that circumscribe nodular areas without altering lobular architecture. There was no necrosis, cholestasis, or significant inflammation. Histopathological diagnosis: congenital hepatic fibrosis (Figs. 2-4). Ophthalmological examination: hamartomas (light brown pigmented neoformations, well circumscribed, 1-2 mm in diameter) in the anterior surface of the iris (Lisch nodules) characteristic of von Recklinghausen's disease (Fig. 5). The patient evolves favorably with periodical medical follow-up.

Discussion
The diagnosis of CHF may be difficult, especially its differentiation from hepatic cirrhosis, in which, unlike CHF, liver architecture is altered with the formation of regenerative nodules. It may be accompanied by inflammation and cell damage, and ductal-like structures are not observed, which are characteristic of CHF (1,3). Unlike cirrhosis, hepatic function is generally preserved in CHF; it usually has a good prognosis, although there would be an increased risk of developing cholangiocarcinoma and renal insufficiency (5,6).
CHF is related to other diseases with ductal plate malformation, being part of the spectrum of fibropolycystic diseases affecting the liver and kidneys. These are Caroli's disease, von Meyernburg's complexes, and polycystic disease (7,8). In the latter there are alterations in the PKD-1 and PKD-2 genes, which encode polycystein proteins 1 and 2 involved in the differentiation and maturation of renal tubules (9). This group of diseases would present an alteration in the interaction between cells and the extracellular matrix, which would interfere with the process of involution and remodelling of the ductal plate, and would thus induce the alterations observed in these conditions (10,11). Some cases of CHF have been associated with phosphomannose isomerase deficiency, with hypoglycosylation and abnormal remodelling of the ductal plate (12). In numerous malformative syndromes (including those involving the nervous system as is the case with Meckel-Gruber syndrome) ductal plate malformation is observed (13-18).
Von Recklinghausen's disease is an autosomal dominant trait, and approximately 50% of cases are sporadic. Its incidence is 1:3500 births, and it clinically manifests during childhood and adolescence (19). This disease affects tissues derived from the neural crest. It is characterized by the presence of cutaneous pale yellow-brown macules or "café au lait" pigmented spots, usually rounded or ovoid with the major axis parallel to cutaneous nerves. Their color is due to a hyperpigmentation of epidermal basal cells (20). The presence of more than 5 of these spots with a diameter greater than 1.5 cm is pathognomonic for von Recklinghausen's disease (19,21). Neurofibromas and ophthalmological alterations may also be seen, including optic nerve gliomas and iris hamartomas (Lisch nodules) (21,22).
The diagnosis of von Recklinghausen's disease is based on clinical criteria (19). In our case it was based on the presence of "café au lait" spots and "Lisch' nodules. It may be associated with intracraneal and spinal astrocytomas, gastrointestinal (and rarely hepatic) neurofibromas, neuroendocrine tumors (pheochromocytoma, gastrinoma, insulinoma, somatostatinoma), carcinoid tumors, pancreatic adenocarcinoma, melanoma, vascular dysplasia (stenosis of the renal artery), and osteoarticular disorders (scoliosis, cysts, pseudoarthrosis) (19-21,23-26). We have found no description of its association with congenital hepatic fibrosis in the literature.
In Von Recklinghausen's disease there are alterations in the NF-1 gene coding for the neurofibromin protein, which acts as tumor suppressor through the regulation of Ras-MAPK, having a distinctive function during wound healing processes and vascular proliferation, and in the composition of myelin (19,27).
In conclusion, we believe that knowledge on the association between CHF and von Recklinghausen's disease is relevant for a better diagnosis, treatment, and follow up of patients involved.
References
1. Cysts and congenital biliary abnormalities. En: Sherlock S, Dooley J. Diseases of the Liver and Biliary System. 11th ed. Oxford: Blackwell Science; 2002. p. 583-96. [ Links ]
2. Suchy FJ. Anatomy, histology, embriology, developmental anomalies, and pediatric disorders of the biliary tract. En: Feldman M, Friedman LS, Sleisenger MH, editors. Sleisenger and Fordtran's gastrointestinal and liver disease. 7th ed. Philadelphia: Saunders; 2002. p. 1019-42. [ Links ]
3. Childhood liver disease and metabolic disorders. En: Scheuer PJ, Lefkowitch JH, editors. Liver biopsy interpretation. 7th ed. Philadelphia: Elsevier Saunders; 2006. p. 267-97. [ Links ]
4. Witzleben CL, Ruchelli E. Cystic diseases of the liver. En: Zakim D, Boyer TD, editors. Hepatology a Textbook of Liver Disease. 4th ed. Philadelphia: Saunders; 2003. p. 1461-80. [ Links ]
5. Daroca J, Tuthillr R, Reed RJ. Cholangiocarcinoma arising in congenital hepatic fibrosis. Arch Pathol 1975; 99: 592. [ Links ]
6. Álvarez E, Bernard O, Brunelle F, et al. Congenital hepatic fibrosis in children. J Pediatr 1981; 99: 370. [ Links ]
7. Summerfield JA, Nagafuchi Y, Sherlock S, et al. Hepatobiliary fibropolycystic diseases. A clinical and histological review of 51 patients. J Hepatol 1986; 2: 141-56. [ Links ]
8. Desmet VJ. Pathogenesis of ductal plate abnomalities. Mayo Clin Proc 1998; 73: 80-9. [ Links ]
9. Wu G, Somlo S. Molecular genetics and mechanism of autosomal dominant polycystic kidney disease. Molec Genet Metab 2000; 69: 1. [ Links ]
10. Terada J, et al. Normal and abnormal development of the human intrahepatic biliary tree: a review. Tohoku J Exp Med 1997; 181: 19. [ Links ]
11. Jorgensen M. The ductal plate malformation. Acta Pathol Microbiol Scand Suppl 1977; 257: 1. [ Links ]
12. Koning TJ, Nikkels PGJ, Dorland L, et al. Congenital hepatic fibrosis in 3 sibilings with phosphomannose isomerase deficiency. Virchow's Arch 2000; 437: 101-5. [ Links ]
13. Bernstein J. Hepatic involvement in hereditary renal syndromes. Birth defects. Original Article Series 1987; 23: 115. [ Links ]
14. Miranda D, Schinella RA, Finegold MJ. Familial renal dysplasia. Microdissection studies with associated central nervous system and hepatic malformation. Arch Pathol 1972; 93: 483. [ Links ]
15. Blankenberg TA, Ruebner BH, Ellis WG. Pathology of renal and hepatic anomalies in Meckel syndrome. Am J Med Genet 1987; 3: 395. [ Links ]
16. Strayer DS, Kissane J. Dysplasia of the kidneys, liver and pancreas: report of a variant of Ivemark's syndrome. Hum Pathol 1978; 19: 228. [ Links ]
17. Bohm N, Fukuda M, Staudt R, et al. Chondroectodermal dysplasia (Ellis-Van Creveld syndrome) with displasia of renal medulla and bile ducts. Histopathology 1978; 2: 267. [ Links ]
18. Nakamura F, Sasaki H, Kajihara H, et al. Case report: Laurence-Moon-Biedl syndrome accompanied by congenital hepatic fibrosis. J Gastroenterol Hepatol 1990; 5: 206. [ Links ]
19. Barkovich AJ, Kuzniecky RI. Congenital, developmental, and neurocutaneous disorders. En: Goldman L, Ausiello D. Cecil Textbook of Medicine. 22nd ed. Philadelphia: Saunders; 2004. p. 2359-63. [ Links ]
20. Neurofibromatosis de Recklinghausen. En: Heide-Marie Heinz. Dermatología. Neckarsulm: Jungjohann. Maguncia; 1983. p. 19-21. [ Links ]
21. Genetic disorders. En: Robbins SL, Cotran RS, editors. Pathologic basis of disease. 2nd ed. Philadelphia: Saunders: Philadelphia. Second Edition 1979; 213-261. [ Links ]
22. Fay A, Jakobiec FA. Diseases of the Visual System. En Goldman L, Ausiello D. Cecil Textbook of Medicine. 22nd ed. Philadelphia: Saunders; 2004. p. 2406-20. [ Links ]
23. Rutgeerts P, Hendricks H, Geboes K, et al. Involvement of the upper digestive tract by systemic neurofibromatosis. Gastrointest Endosc 1981; 27: 22. [ Links ]
24. Guzmán Toro F, Hinestroza D, Colmenares D. Von Recklinghausen disease and hepatic neurofibromatosis. Gen 1995; 49: 303-6. [ Links ]
25. Jensen RT, Norton JA. Pancreatic endocrine Tumors. En: Feldman M, Friedman LS, Sleisenger MH, editors. Sleisenger and Fordtran's gastrintestinal and liver disease. 7th ed. Philadelphia: Saunders; 2002. p. 988-1016. [ Links ]
26. Guillot B, Dalac S, Delaunay M, et al. Cutaneous malignant melanoma and neurofibromatosis type 1. Melanoma Res 2004; 14: 159-63. [ Links ]
27. Koivunen J, Karvonen SL, Yla-Outinen H, et al. NF1 tumor suppressor in epidermal wound healing with special focus on wound healing in patients with type 1 neurofibromatosis. Arch Dermatol Res 2005; Apr 27; (Epub ahead of print). [ Links ]
![]() Correspondence:
Correspondence:
Oliver A. Jorge.
Videla Castillo 1996.
(5500) Mendoza, Argentina.
e-mail: oliverjorge@lanet.com.ar
Recibido: 23-03-06.
Aceptado: 04-04-06.

