










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.99 no.2 Madrid feb. 2007
IMÁGENES EN PATOLOGÍA DIGESTIVA
Angiomatosis mesentérica masiva y hemorragia digestiva baja en paciente con síndrome de Klippel-Trenaunay-Weber
Massive mesenteric angiomatosis and low digestive hemorrhage in a patient with Klippel-Trenaunay-Weber syndrome
J. M. Álamo Martínez, C. Bernal Bellido, M. Socas Macías, J. García-Moreno, J. M. Suárez Grau y A. Galindo Galindo
Servicio de Cirugía General y Digestiva. Hospital Virgen del Rocío. Sevilla
Paciente de 45 años, sin diagnóstico previo, que acude a Urgencias por hemorragia digestiva baja. El hemograma muestra hemoglobina de 8,6, plaquetopenia de 80.000, y signos de coagulación intravascular diseminada con unos dímeros D de 2000. Se objetiva en la exploración física varicosidades peneanas e hipertrofia asimétrica de los miembros (Fig. 1). La colonoscopia y el estudio angiográfico muestran una angiomatosis mesentérica masiva (Fig. 2).


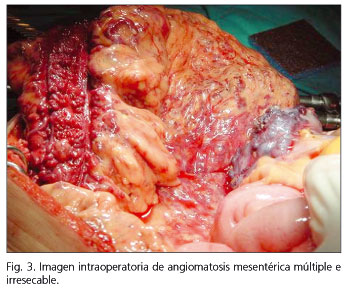
Tras decidirse tratamiento quirúrgico, la laparotomía exploradora confirma los hallazgos descritos, objetivó que existía una angiomatosis mesentérica y mesocólica total y masiva que incluía tanto a vasos iliacos internos y externos, de forma que el proceso era irresecable (Fig. 3), falleciendo el paciente a los pocos meses del diagnóstico por hemorragia intestinal masiva.
El cuadro que presentaba este paciente correspondía con síndrome de Klippel-Trenaunay-Weber con angiomatosis mesentérica y síndrome de coagulación intravascular diseminada asociada a angiomatosis (síndrome de Kassabach-Merrit).
Discusión
El síndrome de Klippel-Trenaunay es una alteración mesodérmica congénita poco frecuente y constituido básicamente por la tríada de hemangiomas, varicosidades e hipertrofia asimétrica de los miembros secundaria a la vascularización anómala congénita. Originalmente fue descrito en 1900, por Klippel y Trenaunay. En 1907, Weber añadió al síndrome la fístula arteriovenosa, una alteración poco frecuente (1). Las alteraciones en el desarrollo de la capa germinal mesoblástica afectan las estructuras angioblásticas, linfoblásticas y osteoblásticas, y determinan que aparezca un número ilimitado de asociaciones malformativas (2-4).
Este síndrome es una entidad clínica rara en el que existen alteraciones vasculares de diferentes localizaciones (5). Las lesiones son muy variadas e incluyen los siguientes tipos: hemangiomas capilares y cavernosos, flebectasia y varicosidades (6). La angiomatosis intracraneal, gastrointestinal, urinaria o broncopulmonar es rara, más aún de forma masiva.
En los miembros ocasionalmente se observan fístulas arteriovenosas (7), anomalías linfangiomatosas y atrofia. Asimismo, macrodactilia (8), crecimiento desproporcionado de los dedos, sindactilia, polidactilia y oligodactilia.
Algunos enfermos presentan hemorragia rectal por varices colónicas, pudiendo existir anemia crónica (5). La hemorragia digestiva baja debe tratarse de forma inicial mediante hemostasia endoscópica, reservándose la resección quirúrgica del tramo afecto cuando fracasa la misma, y en aquellos casos de angiodisplasia masiva (9).
El paciente descrito presentaba no sólo estos estigmas corporales sino que la angiomatosis se extiende a la totalidad de la vascularización del tracto gastrointestinal y colónico, añadiéndose además la particularidad del síndrome de Kassabach-Merrit añadido consecuente con la gran angiomatosis subyacente.
El diagnóstico puede hacerse visualmente cuando las alteraciones sean evidentes. No obstante, los pacientes deberán ser evaluados utilizando una estrategia de imágenes no invasivas (1), que incluya ecografía y Doppler de color, TC y RM. La venografía estaría indicada para evaluar el tratamiento quirúrgico, y la arteriografía cuando se sospeche una fístula arteriovenosa. La angiorresonancia con gadolinio permite una mayor precisión en el diagnóstico por imagen. En relación con el diagnóstico diferencial, las malformaciones vasculares pueden ser una manifestación primaria de otros trastornos esporádicos o hereditarios que no deben confundirse con el síndrome de Klippel-Trenaunay, como los síndromes de Maffucci, Gorham, Bannayan, Riley-Smith, Solomon, Proteus y otros (10).
La mortalidad del SKT es del 1%, aproximadamente, pero por el contrario la morbilidad es alta debida a las complicaciones que se presentan, como sangrado y secuestro plaquetario, hemorragias internas, tromboflebitis, celulitis aséptica, fallo cardiaco en caso de fístulas múltiples o de gran tamaño asociadas, bacteriemias, osteomielitis y predisposición a las fracturas (11).
El disturbio genético origen de esta enfermedad está en continuo estudio. En 2004, Tian y cols. (12) describieron dos defectos genéticos en una proteína asociada a la angiogénesis, la VG5Q. Esta mutación, denominada E133K y encontrada únicamente en un 4% de los pacientes, hace que este péptido se "hiperactive", estimulando la angiogénesis de una forma más intensa que la proteína VG5Q normal. De todas formas, el origen genético de esta mutación continúa sin ser claro, habiéndose encontrado tan sólo una traslocación cromosómica en un paciente.
Bibliografía
1. Berry SA, Peterson C, Mize W, Bloom K, Zachary CH, Blasco P, et al. Klippel-Trenaunay syndrome. Am J Med Genet 1998; 79: 319-26.
2. Al-Salman MM. Klippel-Trenaunay syndrome: Clinical features, complications and management. Surg Today 1997; 27 (8): 735-40-2.
3. Linge C, et al. TMJ morphology and function in a patient with Klippel-Trenaunay syndrome. J Orofac Orthop 2000; 61 (3): 217-21.
4. Coria F, Berciano J. Occipito-cervical dysplasia. Med Clin (Barc) 1985; 84 (5): 199-205.
5. Garteiz D, et al. Rectorrhagia as complication of Klippel-Trenaunay syndrome. Rev Gastroenterol Mex 1999; 64 (4): 181-5.
6. Jacob AG, et al. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clinic Proc 1998; 73 (1): 28-36.
7. Irlbacher K, et al. Variant of Klippel-Trenaunay syndrome. Rontgenpraxis 2000; 52 (9): 312-16.
8. Krengel S, et al. Macrodactyly: report of eight cases and review of the literature. Pediatr Dermatol 2000; 17 (4): 270-6.
9. Duque JM, et al. Colonic involvement in the Klippel-Trenaunay-Weber syndrome. Rev Esp Enferm Dig 2000; 92 (1): 44-5.
10. Silverman RA. Hemangiomas y malformaciones vasculares. Pediatr Clin North Am (ed. esp.) 1991; 38: 835-55.
11. Jacob AG, Driscoll AJ, Shaughnessy WJ, Starson AW, Clay RP, Glovictki P. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clin Proc 1998; 73: 28-36.
12. Tian XL, Kadaba R, You SA, et al. Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature 2004; 427: 640-5.

