










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.100 no.4 Madrid abr. 2008
Expresión génica en pacientes obesos con enfermedad hepática por depósito de grasa
Gene expression in obese patients with non-alcoholic steatohepatitis
A. Cayón, J. Crespo, A. R. Guerra y F. Pons-Romero
Servicio de Aparato Digestivo. Hospital Universitario "Marqués de Valdecilla". Santander
Dirección para correspondencia
RESUMEN
La fisiopatología de la enfermedad hepática por depósito de grasa sólo se conoce de forma parcial. En este trabajo hemos analizado la expresión génica intrahepática de citoquinas, quimioquinas, receptores celulares, factores de crecimiento, transductores de señales intracelulares y proteínas de comunicación extracelular en el tejido hepático de sujetos obesos con y sin esteatohepatitis no alcohólica, en un intento de determinar un perfil de expresión génica asociado a las formas severas de la esteatohepatitis no alcohólica (EHNA).
Se analizó un grupo de 38 pacientes obesos con un IMC > 35, que fueron sometidos a cirugía bariátrica. La expresión génica intrahepática se determinó en el tejido hepático dividiendo a los pacientes en tres grupos: a) pacientes obesos sin datos histológicos sugestivos de EHNA (n = 12); b) pacientes con EHNA sin fibrosis (n = 13); y c) pacientes con EHNA y fibrosis (n = 13). Se consideró que existía una sobreexpresión génica cuando la diferencia en la expresión era, al menos, de dos veces con respecto al grupo control. Los resultados se confirmaron mediante PCR en tiempo real. Se detectó una expresión diferencial de 14 genes (10 sobreexpresados y 4 infraexpresados). Los genes sobreexpresados incluyeron prohibitina, TNF, TNF RI (p55), MCSF, R2-TRAIL, TGF-b1, CTGF, FGF, VEGF, BIGH3 y ObRb. La expresión de los genes insulin growth factor-1, insulin growth factor-2, interleuquina-2 y tyrosine-receptor fue menor que en el grupo control.
En conclusión:
1. Los pacientes obesos con EHNA sin fibrosis muestran una sobreexpresión de genes proinflamatorios y proapoptóticos. En los pacientes con EHNA y fibrosis, se observa, además, una sobreexpresión de genes profibrogénicos, incluyendo el gen del receptor de la leptina.
2. La expresión de prohibitiva en los pacientes con EHNA, tanto con fibrosis como sin fibrosis, fue superior que en los controles, lo que sugiere una disfunción mitocondrial en los pacientes con EHNA.
Palabras clave: Esteatohepatitis no alcohólica. Enfermedad hepática por depósito de grasa. Obesidad mórbida.
ABSTRACT
Although the molecular basis for the pathophysiology of non-alcoholic steatohepatitis (NASH) is poorly understood, we evaluate the hepatic gene expression of cytokines, chemokines, cell receptors, growth factors, intracellular transducers and extracellular communication proteins in liver tissue of obese patients (with and without NASH), and we determine the specific intrahepatic gene expression profiles associated with histological severe NASH.
Thirty-eight obese patients with BMI > 35 were analyzed, who underwent bariatric surgery. Biopsy specimen samples were snap-frozen in liquid nitrogen. Hepatic gene expression was determined in liver biopsy specimens from 3 groups: a) obese patients without NASH (n = 12); b) patients with NASH without fibrosis (n = 13); and c) patients with NASH and fibrosis (n = 13). Genes were considered to be expressed differentially in NASH only if there was a greater than 2-fold difference in abundance of mRNA when compared with each of the control group. These results were confirmed by real-time PCR. Fourteen genes were differentially expressed (10 overexpressed and 4 underexpressed) in patients with NASH. Genes that were significantly overexpressed included prohibitin, TNF, TNF RI (p55), MCSF, R2-TRAIL, b1-CTGF, FGF, VEGF, and BIGH3OBR. Insulin growth factor-1, insulin growth factor-2, interleukin-2 and tyrosine-receptor were underexpressed in NASH patients.
In conclusion:
1. The obese patients with NASH without fibrosis show an overexpression of proinflammatory and proapoptotic genes. Also, the NASH patients with fibrosis show an overexpression of fibrogenic genes, including the leptin receptor Ob-Rb.
2. The up-regulated gene expression of prohibitin suggests mitochondrial dysfunction in NASH patients.
Key words: Non-alcoholic steatohepatitis. Non-alcoholic fatty liver disease. Morbid obesity.
Introducción
La enfermedad hepática por depósito de grasa no alcohólica (NAFLD) abarca un amplio espectro de patología hepática, desde la esteatosis simple a una esteatosis con inflamación, frecuentemente asociada a la fibrosis, llamada esteatohepatitis no alcohólica (NASH), la cual puede progresar a cirrosis (1-4). Actualmente, se acepta la teoría del doble impacto en la patogenia de la EHNA. La esteatosis, consecuencia de la resistencia a la insulina en la mayoría de los pacientes, constituye el primer impacto. Posteriormente, la lipoperoxidación y el estrés oxidativo secundario son capaces de inducir distintas vías implicadas en la secreción de citoquinas proapoptóticas, proinflamatorias y profibrogénicas (5-8). En la patología del NASH se han implicado múltiples sistemas, entre los que se encuentran los relacionados con la resistencia a la insulina, la actividad mitocondrial, el estrés oxidativo y aquellos implicados en el desarrollo de las respuestas inflamatoria y fibrogénica (9-14).
Nuestro grupo de trabajo ha demostrado con anterioridad la implicación del TNF-α, TGF-β1, adiponectina y leptina en la patogenia del NASH. En síntesis, objetivamos un incremento en la expresión del ARNm del TNF-α en tejido hepático y tejido adiposo de pacientes obesos con NASH y un incremento en la expresión del TGF-β1 en pacientes con NASH y fibrosis (15,16). Por otro lado, describimos una marcada disminución de la expresión de la adiponectina y del receptor II de la adiponectina en el tejido hepático de los pacientes con NASH con respecto a un grupo control formado exclusivamente por sujetos con esteatosis hepática (17). Por último, hemos visto que la sobreexpresión del receptor de la leptina está asociada con la fibrosis hepática (resultados no publicados). Sin embargo, otros múltiples genes potencialmente implicados en la patogenia de la enfermedad no se han tipificado con exactitud (18,19).
El objetivo de este estudio ha sido analizar mediante macroarrays de ADNc la expresión génica intrahepática de distintas citoquinas, receptores de citoquinas, quimioquinas, factores de crecimiento y trasductores de señales intracelulares e intercelulares implicados en la fibrosis e inflamación en los pacientes obesos con EHNA. Posteriormente, hemos intentado identificar un patrón de expresión diferencial de los distintos genes implicados en los sujetos con EHNA en función de la existencia de esteatosis, inflamación y fibrosis.
Material y métodos
Pacientes
Se incluyeron 38 pacientes obesos sometidos a cirugía bariátrica en el Hospital Universitario "Marqués de Valdecilla" entre septiembre del año 1999 y enero de 2002. El índice de masa corporal (IMC) era de 46,8 ± 7,8 kg/m2. A todos los pacientes se les efectuó una historia clínica y una exploración física completas; asimismo, en todos los casos se practicaron determinaciones analíticas y, durante el acto quirúrgico, se obtuvo una biopsia hepática, previo consentimiento informado. Se practicaron los siguientes criterios de exclusión:
1. Consumo de alcohol diario superior a 30 gramos en los varones o 20 gramos en las mujeres. La historia de la ausencia de consumo de alcohol fue confirmada por, al menos, dos familiares del paciente.
2. Consumo de fármacos capaces de inducir hepatotoxicidad, incluyendo corticoides, amiodarona, tamoxifeno o tetraciclinas.
3. Presencia de cualquier otra causa potencial de enfermedad hepática. De hecho, todos los pacientes fueron HBsAg y anti-VHC negativos. No se objetivaron títulos significativos de anticuerpos antimitocondriales, antinucleares o antimúsculo liso.
Determinaciones analíticas
Se efectuaron las siguientes determinaciones analíticas: glucosa, creatinina, colesterol, triglicéridos, bilirrubina total, aspartato aminotransferasa (AST), alanina aminotransferasa (ALT), gamma glutamiltransferasa (GGT), albúmina y proteínas totales mediante una autoanalizador automatizado. Asimismo, se determinaron HBsAg, anti-VHC, autoanticuerpos, metabolismo del hierro, ceruloplasmina y alfa 1 antitripsina.
Estudio histológico
Se procedió a la realización de una biopsia hepática mediante aguja tru-cut en todos los pacientes durante la intervención quirúrgica. Se obtuvo el consentimiento informado en todos los casos antes de la misma. Este estudio fue efectuado de acuerdo con la Declaración de Helsinki. Un tercio del material histológico obtenido se introdujo de forma inmediata en nitrógeno líquido y se guardó a -80 ºC hasta su posterior utilización y el resto fue utilizado para el diagnóstico histológico. Un único patólogo (MM) interpretó todas las biopsias hepáticas antes de la realización de los estudios de expresión génica. El tejido hepático se fijó con formalina al 10% y posteriormente se incluyó en parafina. Las biopsias se procesaron de forma rutinaria en cortes de 4 μm y se tiñeron con hematoxilina-eosina. Se utilizó la clasificación propuesta por Brunt y cols. (20,21) para el estadiaje de la lesión hepática.
Aislamiento del ARN
El ARN total se extrajo del tejido hepático mediante el método del guanidinium phenol chloroform usando el TRIzol® (GIBCO BRL, Gaithersburg, Maryland, Estados Unidos) de acuerdo con las instrucciones del fabricante. Este protocolo se basó en el método previamente descrito por Chomezynski y Sacchi (22). La concentración de ARN se valoró mediante espectroscopia y la integridad del ARN se confirmó mediante electroforesis en gel de agarosa no desnaturalizante.
Expresión génica mediante macroarrays
Se efectuó un estudio de la expresión génica mediante macroarrays en 38 pacientes. Estos se dividieron en 3 grupos: 13 pacientes con NASH y fibrosis hepática, 13 pacientes con datos histológicos sugestivos de NASH sin fibrosis y 12 sujetos con esteatosis leve sin inflamación ni fibrosis (grupo control). Para determinar el perfil de expresión génica intrahepática, se analizó la abundancia relativa de ARNm usando una tecnología de macroarrays con oligonucleótidos de alta densidad con sondas para 268 genes (AtlasTM human cytokine array, Clontech).
El ARN total (40 µg, 1 µg/µl) se incubó a 37 ºC durante 30 min con Dnase I, libre de Rnase (Roche, Germany). Se efectuó una doble extracción: fenol, cloroformo y se resuspendió en 15 µl de agua libre de Rnase.
La hibridación del macroarray se efectuó usando 5 mg del ARN hepático total. Cinco microgramos de ARN se retrotranscribieron mediante un CDS primer mix (Clontech) en presencia de 32P dATPs (3.000 Ci/mmol, 10 µCi/µl, Amersham). Las sondas se purificaron mediante un kit de purificación de productos de PCR (Atlas Nucleospin® Extraction Kit, Clontech) de acuerdo con las instrucciones del fabricante.
Las muestras del ADNc se hibridaron durante toda la noche con agitación continua a 68 ºC (BD Express Hyb, Clontech). Posteriormente se procedió a dos lavados consecutivos (2 XSSC, 1% SDS y 0,1 XSSC, 0,5% SDS respectivamente).
La señal se detectó tras exponer la membrana a -70 ºC durante dos y cinco días. La intensidad de la luminiscescencia se analizó mediante el software de Bio-Rad Quantity OneTM (Bio-Rad Laboratories, California, Estados Unidos).
Para identificar la expresión diferencial de los genes asociados con las formas progresivas de EHNA se procedió a un análisis en dos fases. Inicialmente, la expresión de cada array se normalizó usando varios genes marcadores (ubiquitina, GAPDH, HLA C-4, β-actin, tubulina, alfa 1, proteína ribosómica 60S L13A y proteína ribosómica 40S S9). Posteriormente, el porcentaje de cambio de la expresión de cada gen fue calculado mediante las fórmulas: intensidad en sujetos con EHNA/intensidad en obesos sin EHNA; e intensidad en sujetos con EHNA y fibrosis/intensidad en sujetos con EHNA sin fibrosis. Se consideró que existía una expresión diferencial de los genes cuando estos se expresaban, al menos, dos veces más o dos veces menos que los controles.
PCR en tiempo real
Para validar los resultados de la hibridación, se efectuó una PCR en tiempo real de los genes cuya expresión estaba aumentada o disminuida en el macroarray. Las sondas y primers utilizados para la Taqman PCR (QPCR) de pro-hibitina, TNFRSF10B, ILF-1, ILF2, VEGF, FGF, CTGF, CSF1, TGF y r18S se obtuvieron de Applied Biosys-tems (Foster City, California, EE.UU.): prohibitina (Hs00855044), TNFRSF10B (Hs00187196), ILF-1 (Hs00153126), ILF2 (Hs001712554), VEGF (Hs00173626), FGF (Hs00265254), CTGF (Hs00170014), MCSF (Hs00174164), TGFβ1 (Hs00165908) y r18S (Hs). Los ensayos TaqMan se realizaron en un volumen total de 20 ml. En todos los ensayos se incluyó un control sin molde. La expresión se normalizó en función de la expresión del ARN ribosómico 18S. Los ensayos se efectuaron en duplicado (MicroAmp 96-well reacción plates, Applied Biosystem) en un AbiPrism 7000 Sequence Detector System (Applied Biosystem). Las condiciones de amplificación fueron: 2 min a 50 ºC y 10 min a 95 ºC seguidas por 40 ciclos de 15 s a 95 ºC y 1 min a 60 ºC. Los resultados se analizaron mediante el software de ABI Prism 7000 SDS.
Análisis estadístico
Se expresaron los datos como media ± SEM. Las diferencias entre los grupos se calcularon usando la t de Student para muestras independientes. Se consideró que existía una diferencia significativa cuando la p era menor de 0,05. Los coeficientes de correlación se calcularon mediante el método de Pearson. Se utilizó el paquete informático SPSS (versión 11.0).
Resultados
Características clínicas e histológicas
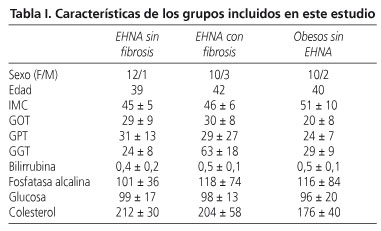
De los 38 pacientes incluidos en el estudio, 13 pacientes tenían NASH con fibrosis, 13 pacientes tenían NASH sin fibrosis y 12 sujetos tenían una esteatosis leve sin inflamación ni fibrosis, este grupo lo hemos considerado el control interno. Todos los pacientes, en los tres grupos, mostraban similares características biomédicas. Los detalles de las características demográficas, clínicas y analíticas son mostrados en la tabla I.
Hemos estudiado el grado de esteatosis, el grado de inflamación y el estadio de fibrosis en todos los pacientes. Las características histológicas están representadas en la tabla II.

Macroarrays de expresión génica
Mediante este ensayo, hemos detectado genes sobreexpresados y genes inhibidos en todos los pacientes con NASH respecto al grupo control de sujetos obesos sin NASH (Tabla III).
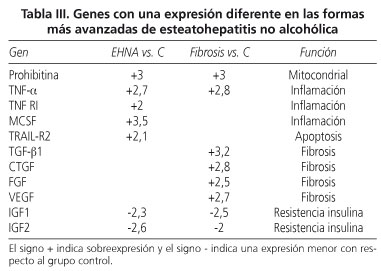
Los pacientes con NASH con inflamación sin fibrosis respecto al grupo control de pacientes obesos sin NASH muestran 5 genes sobreexpresados y dos genes inhibidos. Los genes sobreexpresados fueron: a) tres genes relacionados con la respuesta inflamatoria: tumor necrosis growth factor alfa (TNF-α, +2,7), tumor necrosis growth factor receptor I (TNFRI, +2), y macrophage colony-stimulating factor (M-CSF, +3,5); b) 1 gen relacionado con la respuesta apoptótica: TNF-related apoptosis-inducing ligand receptor 2 (TRAIL-R2, +2,1); y c) un gen involucrado en la función mitocondrial: prohibitina (PHB, +3). Los genes inhibidos en estos pacientes respecto a los pacientes obesos sin daño hepático son genes involucrados en la resistencia a la insulina: insulin-like growth factor I (IGFI, -2,3) e insulin-like growth factor II (IGFII, -2,6).
Los pacientes con NASH con fibrosis respecto al grupo de pacientes sin NASH, control, muestran 6 genes sobreexpresados y un gen inhibido. Los genes sobreexpresados son: a) un gen involucrado en la respuesta inflamatoria: TNF-α (+2,8); b) 4 genes implicados en la respuesta fibrótica: transforming growth factor β (TGFβ1, +3,2), fibroblast growth factor (FGF, +2,5), vascular endothelial growth factor (VEGF, +2,7) y connective tissue growth factor (CTGF, +2,8); y c) un gen implicado en la función mitocondrial: PHB, +3. El gen inhibido fue el IGFI (-2,5), relacionado con la resistencia a la insulina.
PCR en tiempo real
El estudio de comprobación de la expresión génica de los resultados obtenidos en el array se realizó por PCR cuantitativa a tiempo real (QPCR), normalizando los resultados de las citoquinas con respecto al estándar interno, el gen ribosomal 18S. En los pacientes con NASH sin fibrosis con respecto a los pacientes sin NASH la QPCR confirmó la sobreexpresión del TRAIL-R2 , MCSF y la prohibitina; y la inhibición del IGFI e IGFII: TRAIL-R2 (3,1 ± 2 vs. 1,6 ± 1,4; p < 0,04), MCSF (1,6 ± 1,2 vs. 0,8 ± 0,4; p < 0,008), prohibitin (1,3 ± 1,1 vs. 0,8 ± 0,4; p < 0,05), IGFI (0,6 ± 0,5 vs. 1,1 ± 0,4; p < 0,03), IGFII (0,6 ± 0,8 vs. 1,2 ± 1,4; p < 0,02). El incremento en la expresión génica hepática del TNF-α ARNm y TNFRI ARNm en los pacientes con NASH respecto a los pacientes obesos sin NASH fue determinado previamente por PCR semicuantitativa (9).
En los pacientes con NASH sin fibrosis respecto a los sujetos sin NASH la QPCR confirmó la sobreexpresión del VEGF, CTGF, TGFβ1 y prohibitina; y la inhibición del ILGI: VEGF (1,8 ± 2,3 vs. 0,7 ± 1,2; p < 0,05), CTGF (2,2 ± 1,2 vs. 1,1 ± 0,6; p < 0,011), TGFβ1 (2,66 ± 1,32 vs. 1,3 ± 0,9; p < 0,002), PHB (1,8 ± 1,4 vs. 0,9 ± 0,6; p < 0,014), ILFI (0,4 ± 0,4 vs. 0,9 ± 0,5; p < 0,01). El FGF mostró una expresión similar en todos los pacientes con NASH y fibrosis, NASH sin fibrosis y pacientes sin NASH: (1,2 ± 0,6 vs. 1,2 ± 0,7; p = ns). La sobreexpresión del TNF-α ARNm en los pacientes con EHNA y fibrosis respecto a los pacientes sin EHNA fue determinada previamente por PCR semicuantitativa (10).
Discusión
Hemos realizado unos análisis de expresión génica mediante macroarrays, para intentar establecer un patrón de expresión de distintos genes implicados en la inflamación y en la fibrosis en los pacientes con NASH. Hemos comparado la expresión génica de 268 genes de citoquinas, receptores de citoquinas, trasductores de señales intracelulares e intercelulares y factores de crecimiento en los pacientes con NASH con y sin fibrosis respecto a los pacientes obesos sin daño hepático.
En este trabajo hemos encontrado que todos los pacientes obesos con NASH respecto a los sujetos obesos sin NASH (grupo control) muestran una clara sobreexpresión de los genes que codifican para la proteína mitocondrial prohibitina y para la citoquina inflamatoria TNF-α. La prohibitina es una chaperona que interviene en el ensamblaje de la cadena respiratoria mitocondrial (23-26). La prohibitiva contribuye a proteger la cadena respiratoria mitocondrial de su degradación por la mAAA-proteasa, estando vinculada la expresión anormal de esta proteína en los pacientes obesos con el riesgo de desarrollo de esteatohepatitis. En la EHNA, las subunidades de esta cadena están disminuidas, lo que podría explicar el descenso en las subunidades de la cadena respiratoria (27). Sin embargo, otros autores han descrito en tumores hepáticos primarios una sobreexpresión de la prohibitina en respuesta al estrés metabólico (28). Nuestros resultados sugieren que una alteración en la expresión de la prohibitina puede perjudicar la organización funcional y nativa de la cadena respiratoria, comprometiendo la funcionalidad de la mitocondria. En este sentido, un aumento de la expresión génica podría ser compatible con un descenso en la cuantía proteica o en la actividad proteica, siendo el incremento de la expresión génica un intento de compensación de la disminución de la actividad proteica.
Además, se ha demostrado que la disfunción mitocondrial juega un papel central en la patogénesis del NASH (14,29), la resistencia periférica a la insulina incrementa la beta-oxidación de los ácidos grasos y que el estrés oxidativo está presente tanto en la esteatosis como en la estea-tohepatitis, pero que el NASH sólo se asocia con defectos en la estructura mitocondrial (10).
Los pacientes obesos con NASH que presentan un mayor grado de inflamación en la biopsia hepática muestran una marcada sobreexpresión de los genes que codifican para el TNFRI, el MCSF y el TRAIL-R2 con respecto al grupo control (sujetos con esteatosis hepática simple). La sobreexpresión del TNFRI, al igual que el TNF-α, ya había sido encontrada previamente por ensayos de PCR semicuantitativa. El MCSF es una citoquina que contribuye al incremento del número de macrófagos y por tanto a la inflamación (30,31). Previamente, se había mostrado que los valores séricos del MCSF estaban incrementados con la necrosis e inflamación hepática en pacientes con hepatitis aguda y en pacientes con cirrosis (32). También se ha visto un incremento en sangre periférica total del IL-1 beta, TNF-α e IL-6, correlacionando con el MCSF y con la inflamación hepática (33). Nuestros resultados revelan un incremento en la expresión del ARNm de ambas citoquinas MCSF y TNF-α en el hígado, sugiriendo la activación de la vía de cascada de citoquinas, resultando la proliferación de macrófagos in vivo en el hígado. El otro gen sobreexpresado en los pacientes con NASH con mayor grado de inflamación respecto a los controles fue el TRAIL-R2. El TNF-related apoptosis-inducing ligand (TRAIL) es un miembro de la superfamilia del TNF, el cual induce apoptosis en una variedad de células cancerosas con muy pocos o ningún efecto en células normales (34). Previamente, usando un modelo de hepatitis por adenovirus, otros autores pudieron demostrar que la apoptosis en los hepatocitos infectados por el virus está mediada por el TRAIL-R2 y el TRAIL (35). En este trabajo, nosotros mostramos una sobreexpresión del TRAIL-R2 en los pacientes con NASH, sugiriendo que el sistema del TRAIL es un importante mediador de la apoptosis en los pacientes con NASH.
Los pacientes con NASH y fibrosis con respecto a los controles muestran además sobreexpresión de 4 genes implicados en la fibrosis hepática: TGFβ1, CTGF, FGF y VEGF. Previamente, se había mostrado un incremento en la producción del TGFβ1 en la fibrosis de otras enfermedades hepáticas, incluyendo la hepatitis crónica por VHC, la hepatopatía alcohólica y la hepatitis autoinmune (36-38). En este sentido, recientemente, nosotros hemos encontrado sobreexpresión del TGFβ1 en el tejido hepático de NASH de pacientes con fibrosis por análisis de PCR en tiempo real, correlacionando con la expresión del receptor de la leptina, sugiriendo una activación del TGFβ1 debido a la acción de la leptina (15) y contribuyendo por tanto a la fibrosis en los pacientes con NASH. El CTGF es una proteína involucrada en la regulación de la proliferación celular, diferenciación y embriogénesis (39,40). También se ha descrito su implicación en la patogenia de múltiples enfermedades caracterizadas por la fibrosis tisular (41,42). Otros estudios han mostrado que el CTGF está sobreexpresado en el hígado durante el curso del NASH, tanto en biopsias hepáticas humanas como en modelos experimentales de obesidad y diabetes (43). Además, estudios in vitro muestran que la glucosa y la insulina inducen un incremento significativo en el ARNm CTGF y la proteína por las células estrelladas hepáticas (HSC) y el CTGF regula al alza varios componentes de la matriz extracelular (44). Se ha sugerido que la continua presencia del TNF-α y del TGFβ1 puede ser necesaria para estimular la síntesis de proteínas de la matriz y para mantener activadas las células estrelladas hepáticas (HSC), cuando el daño hepático está generado (45,46). Además, ambas citoquinas pueden estimular la expresión del CTGF en las células estrelladas hepáticas y regular positivamente la síntesis del inhibidor de proteasas en las células estrelladas activadas, la cual reduce la degradación de la matriz, contribuyendo a la fibrosis del hígado (47,48). Haber encontrado sobreexpresión de estos tres genes, TNF-α, TGFβ1 y CTGF, en los pacientes con EHNA con fibrosis, sugiere que la simultaneidad de la sobreexpresión puede explicar la progresión de la fibrosis en estos sujetos. Por el contrario, cuando la expresión de estas citoquinas no es simultánea, la enfermedad hepática no progresa. Hay otras citoquinas que estimulan la proliferación de las células estrelladas. Estas incluyen la trombina, FGF y VEGF (49). Mediante los análisis de macroarrays de expresión génica y QPCR hemos visto sobreexpresión del VEGF, sin embargo la sobreexpresión del FGF sólo la hemos encontrado en los macroarrays y no en los ensayos en PCR en tiempo real. El FGF se ha visto en modelos experimentales que está sobreexpresado en la fibrosis hepática inducida por concanavilina A, al igual que el TGFβ1 (50,51); sin embargo no hay trabajos que muestren la implicación de estos dos genes (VEGF y FGF) en el desarrollo del NASH y se debería hacer un análisis más detallado de estos genes en la patogenia de NASH.
Los genes inhibidos en los pacientes con NASH con y sin fibrosis con respecto a los pacientes sin daños en el hígado son factores de crecimiento relacionados con la insulina, el IGFI y el IGFII. Estos factores de crecimiento son hormonas anabólicas, marcadores del estatus nutricional y de la función hepática, cuya biodisponibilidad está reducida en la cirrosis del hígado (52-54). Bajos niveles de IGF1 y altos niveles de IGFBP-1 (proteína de unión al IGF1) han sido descritos previamente en asociación con la cirrosis y la resistencia a la insulina (55,56). La insulina es la principal reguladora de la expresión del IGFBP-1 hepático, mediante un descenso en el plasma del IGFBP-1 en respuesta al estado nutricional. Los efectos del incremento en la expresión del IGFBP-1 se deben a un descenso en la disponibilidad intracelular del IGF1 libre y la consecuente resistencia a la insulina (57). Sreekumar fue el primero que documentó una sobreexpresión del IGFBP-1 en los pacientes con NASH sin cirrosis respecto a los pacientes sin daño hepático (58). En este sentido, nosotros encontramos una inhibición del IGF1 en los pacientes con NASH sin cirrosis. Estos resultados sugieren que la esteatohepatitis está asociada con alteraciones en la expresión del IGF1 y otros mediadores de la acción de la insulina. Sin embargo, también cabe la posibilidad de que los genes relacionados con la resistencia a la insulina se expresen de forma diferente en los sujetos obesos a como se expresan en personas totalmente sanas. En este sentido, un trabajo que analizara las diferencias en la expresión génica entre sujetos totalmente sanos, obesos y pacientes obesos con EHNA podría aclarar este aspecto.
En resumen, nuestros resultados revelan que en la progresión del NASH intervienen genes implicados en vías proinflamatorias, proapoptóticas y profibrogénicas y con la resistencia a la insulina. Los pacientes obesos con NASH respecto a los controles muestran activación de citoquinas inflamatorias y apoptóticas e inhibición de genes implicados en la resistencia a la insulina. Los pacientes con NASH y fibrosis respecto a los controles muestran también activación de citoquinas fibróticas. Además la sobreexpresión del gen de la prohibitina sugiere la disfunción mitocondrial en los todos pacientes con NASH con y sin fibrosis.
Agradecimientos
Este trabajo se ha realizado gracias a una beca del programa I + D (SAF 2001/0876), una beca del Fondo de Investigaciones Sanitarias de la Seguridad Social (PI020858) y a los fondos de la Red temática de investigación cooperativa: Estudio de los mecanismos patogénicos de las hepatitis víricas y esteatohepatitis: implicaciones en la terapia farmacológica (G03/015).
Bibliografía
1. Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis 2004; 24: 3-20. [ Links ]
2. Bacon BR, Farahvash MJ, Janney CG, Neusch-Wander-Tetri BA. Nonalcoholic steatohepatitis: An expanded clinical entity. Gastroenterol 1994; 107: 1103-9. [ Links ]
3. Harrison SA, Di Bisceglie AM. Advances in the understanding and treatment of nonalcoholic fatty liver disease. Drugs 2003: 63: 2379-94. [ Links ]
4. Poves Prim I, Macías GJ, Cabrera Fraga M, Situ L, Ballesta López C. Calidad de vida en la obesidad mórbida. Rev Esp Enferm Dig 2005; 97: 187-95. [ Links ]
5. Day CP, James OFW. Steatohepatitis: A tale of two "hits"? Gastroenterology 1998; 114: 842-5. [ Links ]
6. Wanless IR, Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: A four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin Liver Dis 2004; 24: 99-106. [ Links ]
7. Choudhury J, Sanyal AJ. Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin Liver Dis 2004; 8: 575-94. [ Links ]
8. Videla LA, Rodrigo R, Araya J, Poniachik J. Oxidative stress and depletion of hepatic long-chain polyunsaturated fatty acids may contribute to nonalcoholic fatty liver disease. Free Radic Biol Med 2004; 37: 1499-507. [ Links ]
9. Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med 1999; 107: 405-55. [ Links ]
10. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterol 2001; 120: 1183-92. [ Links ]
11. Solís Herruzo JA, García Ruiz I, Pérez Carreras M, Muñoz Yagüe MT. Enfermedad grasa del hígado no alcohólica. Desde la resistencia a la insulina a la disfunción mitocondrial. Rev Esp Enferm Dig 2006; 98: 844-74. [ Links ]
12. Romero-Gómez M. Hepatitis C y resistencia a la insulina: esteatosis, fibrosis y no respuesta. Rev Esp Enferm Dig 2006; 98: 605-15. [ Links ]
13. Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 2004; 40: 46-54. [ Links ]
14. Pérez-Carreras M, del Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003; 38: 999-1000. [ Links ]
15. Crespo J, Cayón A, Fernández-Gil P, Hernández Guerra M, Mayorga M, Domínguez-Díez A, et al. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001; 34: 1158-63. [ Links ]
16. Crespo J, Cayón A, Mayorga M, Domínguez-Díez A, Hernández-Guerra M, Fernández-Gil P, et al. Role of transforming growth factor beta (TGF-b) in hepatic fibrosis of non-alcoholic steatohepatitis patients. Madrid: EASL; 2002. [ Links ]
17. Kaser S, Moschen A, Cayón A, Kaser A, Crespo J, Pons-Romero F, et al. Adiponectin and its receptors in nonalcoholic steatohepatitis. Gut 2005; 54: 117-21. [ Links ]
18. Younossi ZM, Baranova A, Ziegler K, Del Giacco L, Schlauch K, Born TL, et al. A genomic and proteomic study of the spectrum of nonalcoholic fatty liver disease. Hepatology 2005; 42: 665-74. [ Links ]
19. Younossi ZM, Gorreta F, Ong JP, Schlauch K, Giacco LD, Elariny H, et al. Hepatic gene expression in patients with obesity-related non-alcoholic steatohepatitis. Liver Int 2005; 25: 760-71. [ Links ]
20. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am J Gastroenterol 1999; 94: 2467-74. [ Links ]
21. Brunt EM. Nonalcoholic steatohepatitis: Definition and pathology. Semin Liver Dis 2001; 21: 3-16. [ Links ]
22. Chomczynski P, Sacchi V. Single step method of RNA isolation by acid guanidium thiocynate-phenol-chloroform extraction. Anal Biochem 1987; 162: 156-9. [ Links ]
23. Nijtmans LG, de Jong L, Artal Sanz M, Coates PJ, Berden JA, Back JW, et al. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J 2000; 19 (11): 2444-51. [ Links ]
24. Kirchman PA, Miceli MV, West RL, Jiang JC, Kim S, Jazwinski SM. Prohibitins and Ras2 protein cooperate in the maintenance of mitochondrial function during yeast aging. Acta Biochim Pol 2003; 50: 1039-56. [ Links ]
25. Nijtmans LG, Artal SM, Grivell LA, Coates PJ. The mitochondrial PHB complex: Roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell Mol Life Sci 2002; 59: 143-55. [ Links ]
26. Coates PJ, Nenutil R, McGregor A, Picksley SM, Crouch DH, Hall PA, et al. Mammalian prohibitin proteins respond to mitochondrial stress and decrease during cellular senescence. Exp Cell Res 2001; 265: 262-73. [ Links ]
27. Santamaría E, Ávila MA, Latasa MU, Rubio A, Martín-Duce A, Lu SC, et al. Functional proteomics of nonalcoholic steatohepatitis: Mitochondrial proteins as targets of S-adenosylmethionine. Proc Natl Acad Sci USA 2003; 100: 3065-70. [ Links ]
28. Suh SK, Hood BL, Kim BJ, Conrads TP, Veenstra TD, Song BJ. Identification of oxidized mitochondrial proteins in alcohol-exposed human hepatoma cells and mouse liver. Proteomics 2004; 4: 3401-12. [ Links ]
29. Fromenty B, Robin MA, Igoudjil A, Mansouri A, Pessayre D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab 2004; 30: 121-38. [ Links ]
30. Miyakawa K, Myint YY, Takahashi K. Effects of recombinant human macrophage colony-stimulating factor on proliferation, differentiation and survival of Kupffer cells in the liver of adult mice. Anal Quant Cytol Histol 1999; 21: 329-35. [ Links ]
31. Pinzani M, Abboud HE, Gesualdo L, Abboud SL. Regulation of macrophagecolony stimulating factor in liver fat-storing cells by peptide growth factors. Am J Physiol 1992; 262 (4 Pt 1): C876-81. [ Links ]
32. Itoh Y, Okanoue T, Ohnishi N, Nishioji K, Sakamoto D, Nagao Y, et al. Hepatic damage induced by transcatheter arterial chemoembolization elevates serum concentrations of macrophage-colony stimulating factor. Liver 1999; 19 (2): 97-103. [ Links ]
33. Itoh Y, Okanoue T, Enjyo F, Sakamoto S, Ohmoto Y, Nirai Y, et al. Serum levels of macrophage colony stimulating factor (M-CSF) in liver disease. J Hepatol 1994; 21 (4): 527-35. [ Links ]
34. Taimr P, Higuchi H, Kocova E, Rippe RA, Friedman S, Gores GJ. Activated stellate cells express the TRAIL receptor-2/death receptor-5 and undergo TRAIL-mediated apoptosis. Hepatology 2003; 37 (1): 87-95. [ Links ]
35. Mundt B, Kuhnel F, Zender L, Paul Y, Tillmann H, Trautwein C, et al. Involvement of TRAIL and its receptors in viral hepatitis. FASEB J 2003; 17 (1): 94-6. [ Links ]
36. Anonni G, Weiner FR, Zern MA. Increased transforming growth factor beta 1 gene expression in human liver disease. J Hepatol 1992; 14: 259-64. [ Links ]
37. Bedossa P, Peltier E, Terris B, Franco D, Poynard T. Transforming growth factor-beta 1 (TGF-beta 1) and TGF-beta 1 receptors in normal, cirrhotic, and neoplastic human livers. Hepatology 1995; 21: 760-6. [ Links ]
38. Kinnman N, Andersson U, Hulterantz R. In situ expression of transforming growth factor beta 1-3, latent transforming growth factor beta binding protein and tumor necrosis factor alpha in liver tissue from patients with chronic hepatitis C. Scand J Gastroenterol 2000; 35: 1294-300. [ Links ]
39. Brigstock DR. The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed family. Endocr Rev 1999; 20: 189-206. [ Links ]
40. Bork P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett 1993; 327: 125-30. [ Links ]
41. Igarashi A, Nashiro K, Kikuchi K, Sato S, Ihn H, Fujimoto M, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disease. J Invest Dermatol 1996; 106: 729-33. [ Links ]
42. Ito Y, Aten J, Bende RJ, Oemar BS, Rabelink TJ, Weening JJ, et al. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 1998; 53: 853-61. [ Links ]
43. Paradis V, Dargere D, Vidaud M, De Gouville, Huet S, Martínez V, et al. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology 1999; 30: 9688-976. [ Links ]
44. Paradis V, Perlemuter G, Bonvoust F, Dargere D, Parfait B, Vidaud M, et al. High glucose and hiperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001; 34: 738-44. [ Links ]
45. Saile Matthes N, Knittel T, Ramadori G. Transforming growth factor beta and tumor necrosis factor alpha inhibit both apoptosis and proliferation of activated rat hepatic stellate cells. Hepatology 1999; 30: 196-202. [ Links ]
46. Knittel T, Muller R, Saile B, Ramadori G. Effect of tumor necrosis factor alpha on proliferation, activation, and protein synthesis of rat hepatic stellate cells. J Hepatol 1997; 27: 1067-80. [ Links ]
47. Liu X, Wu H, Liu F, Huang M, Qiang O, Huang S. Effects of tumor necrosis factor alpha on the expression of connective tissue growth factor in hepatic stellate cells. Zhonghua Gan Zang Bing Za Zhi 2001; 9 (Sup. 1): 15-7. [ Links ]
48. Knittel T, Mehde M, Kobold D, Saile B, Dinter C, Ramadori G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: Regulation by TNF-a and TGF-b1. J Hepatol 1999; 30: 48-60. [ Links ]
49. Rosenbaum J, Blazejewski S, Preaux AM, Mallat A, Dhumeaux D, Mavier P. Fibroblast growth factor 2 and transforming growth factor beta 1 interactions in human liver myofibroblasts. Gastroenterology 1995; 109 (6): 1986-96. [ Links ]
50. Ankoma-Sey V, Matli M, Chang KB, Lalazar A, Donner DB, Wong L, et al. Coordinated induction of VEGF receptors in mesenchymal cell types during rat hepatic wound healing. Oncogene 1998; 17 (1): 115-21. [ Links ]
51. Kimura K, Ando K, Ohnishi H, Ishikawa T, Kakumu S, Takemura M, et al. Immunopathogenesis of hepatic fibrosis in chronic liver injury induced by repeatedly administered concanavalin A. Int Immunol 1999; 11 (9): 1491-500. [ Links ]
52. Castilla-Cortázar I, García M, Muguerza B, Quiroga J, Pérez R, Santidrián S, et al. Hepatoprotective effects of insulin-like growth factor I in rats with carbon tetrachloride-induced cirrhosis. Gastroenterology 1997; 113: 1682-91. [ Links ]
53. Shmueli E, Miell JP, Stewart M, Alberti KG, Record CO. High insulin-like growth factor binding protein 1 levels in cirrhosis: Link with insulin resistance. Hepatology 1996; 24: 127-33. [ Links ]
54. Shmueli E, Record CO, Alberti KG. Insulin independent glucose uptake in cirrhosis. Hepatology 1995; 21: 265-6. [ Links ]
55. Shmueli E, Stewart M, Alberti KG, Record CO. Growth hormone, insulin-like growth factor-1 and insulin resistance in cirrhosis. Hepatology 1994; 19: 32- 8. [ Links ]
56. Lee PD, Jensen MD, Divertie GD, Heiling VJ, Katz HH, Conover CA. Insulinlike growth factor-binding protein-1 response to insulin during suppression of endogenous insulin secretion. Metabolism 1993; 42: 409-14. [ Links ] 57. Wu JC, Daughaday WH, Lee SD, Hsiao TS, Chou CK, Lin HD, et al. Radioimmunoassay of serum IGF-I and IGF-II in patients with chronic liver diseases and hepatocellular carcinoma with or without hypoglycemia. J Lab Clin Med 1988; 112: 589-94. [ Links ] 58. Sreekumar R, Rosado B, Rasmussen D, Charlton M. Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis. Hepatology 2003; 38: 244-51. [ Links ] Recibido: 11-12-07. ![]() Dirección para correspondencia:
Dirección para correspondencia:
Javier Crespo.
Servicio de Aparato Digestivo.
Hospital Universitario "Marqués de Valdecilla", s/n.
39008 Santander.
e-mail: javiercrespo@teleline.es
Aceptado: 29-01-08.

