










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versão impressa ISSN 1130-0108
Rev. esp. enferm. dig. vol.100 no.11 Madrid Nov. 2008
Tumores del estroma gastrointestinal. Estudio retrospectivo de 43 casos
Gastrointestinal stromal tumors - a retrospective study of 43 cases
S. Alberto, P. Sánchez, M. Oliveira, L. Cuesta, F. Gomes, A. Figueiredo, N. Pinheiro y J. Ramos de Deus
Hospital Fernando Fonseca. Lisboa, Portugal
Dirección para correspondencia
RESUMEN
Introducción: los tumores del estroma gastrointestinal (GIST) son poco frecuentes, con una incidencia de 10 a 20 casos por millón de habitantes y año. Aparecen en todo el tubo digestivo, mesenterio o epiplón adyacente; siendo más frecuentes en el estómago (60-70%); también pueden aparecer en el intestino delgado (20-25%), colon y recto (5%) y esófago (< 5%). Su presentación varía desde pequeños nódulos asintomáticos hasta formas más agresivas. Su clasificación se realiza actualmente con base a los criterios de Fletcher.
Objetivo: revisión y caracterización de los casos de GIST observados en nuestro centro durante un periodo de 10 años.
Métodos: estudio retrospectivo de pacientes diagnosticados con GIST (identificados por criterios inmunohistoquímicos) desde enero de 1997 hasta diciembre de 2007 y clasificados por los criterios de Fletcher.
Resultados: se estudiaron 43 pacientes (24 hombres y 19 mujeres), con una edad media de 62,7 años. La mayoría de los GIST encontrados se localizaban en el estómago (n = 20, 46,5%), intestino delgado (n = 18, 41,9%) y en 5 casos se detectaron como metástasis de un tumor oculto. Dieciocho casos fueron asintomáticos. Por los criterios de Fletcher 19 eran casos de alto riesgo, 7 de riesgo intermedio, 12 de bajo riesgo y 5 de riesgo in-determinado. Diez pacientes fallecieron por progresión de la enfermedad y 13 pacientes presentaron metástasis a distancia.
Conclusiones: en nuestra serie, tal como en la literatura, se observa un predominio del sexo masculino y mayor frecuencia de localización gástrica. La supervivencia fue del 42% a los 5 años. La aplicación de los criterios de Fletcher fue consistente con la evolución.
Palabras clave: Tumores estroma gastrointestinal. Criterios de Fletcher. Imatinib. KIT. Leiomiomas.
ABSTRACT
Background: gastrointestinal stromal tumors (GISTs) are rare (10 to 20/million). They exist in the whole digestive system and its surroundings, and are most common in the stomach (70%), followed by the small intestine (20-25%), colon and rectum (5%), and esophagus (< 5%). Their clinical presentation varies from small, incidentally found nodules to large and aggressive tumors. Nowadays GISTs are classified according to Fletcher's classification.
Objective: to review the features of our GIST population.
Methods: a retrospective study of GIST patients identified by immunohistochemical criteria, from 1997 to December 2007, and classified according to Fletcher's criteria.
Results: 43 patients were included (24 men, 19 women) with a mean age of 62.7 years. Gastric GISTs (20 cases, 46.5%), small intestine GISTs (18 cases, 41.9%); in 5 cases metastases of occult tumors were found. Eighteen cases had no symptoms. Tumors were classified according to Fletcher's criteria as high-risk (n = 19), intermediate-risk (n = 7), low-risk (n = 12), and indeterminate-risk (n = 5). Death occurred in 10 patients, and 13 patients had metastatic disease.
Conclusions: our results are in accordance with the world literature, in which a majority of cases are men with gastric tumors. The 5-year survival rate was 42%. Fletcher's criteria were easily applicable criteria and could predict tumor behavior.
Key words: Gastrointestinal stromal tumour. Fletcher's Criteria. Imatinib. KIT. Leiomyomas.
Introducción
Los tumores del estroma gastrointestinal (GIST), aunque poco frecuentes (variando de 0,1% de los tumores malignos del colon hasta 13,9% de los del estómago), son los tumores mesenquimatosos más comunes del tubo digestivo (TD) (1-4). En la década de los 70 se creía que tenían origen en células musculares lisas, siendo entonces clasificados como leiomiomas o leiomiosarcomas (2,4). En 1983, con la llegada de la microscopía electrónica y la inmunohistoquímica se hizo más claro que se trataba de un grupo independiente de lesiones; Mazur y Clark usaron por primera vez el término tumor del estroma para designar a estos tumores mesenquimatosos (2,4).
Los GIST son tumores pleomórficos de células fusiformes, epitelioides y ocasionalmente mesenquimatosas (1,4), con probable origen en las células de Cajal, presentes en las paredes del TD o en las item cell que originan las células de Cajal y las células musculares lisas (4).
El perfil inmunocitoquímico que caracteriza a los GIST empezó a ser descrito con positividad prácticamente sólo para la vimentina. En los años 80 se verificó que el anticuerpo anti-CD 34 era marcador en un gran porcentaje de estos tumores (cerca del 70%). Posteriormente se verificó también positividad para otros anticuerpos como para la actina (20-30%), para la proteína S100 (10%) y < 5% para la desmina (4-6). Finalmente, al final de los años 90, con el uso del CD 117 (c-kIT), se obtuvo un marcador que es positivo en más del 90% de los casos, siendo casi condición sine qua non para el diagnóstico de los mismos (2,4-6).
La proteína KIT (o CD 117) es un receptor transmembrana tirosín-quinasa tipo III cuyo ligando es un factor de item cell (2,4); es también expresada por los mastocitos, item cells de los órganos hematopoyéticos, melanocitos, células germinativas, epitelio del ducto mamario, angiosarcomas, melanomas y seminomas (4). La proteína KIT aparece mutada en 90% de los GIST (2,3). Para los restantes casos que no expresan la mutación en la proteína KIT, es posible buscar la expresión de una mutación en el factor de crecimiento alfa derivado de las plaquetas (PDGFRA-platelet-derved growth factor receptor alpha), descrito en 2003 (2,3).
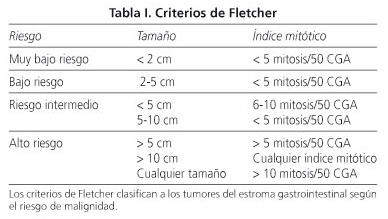
En 2002 fue creada por Fletcher una clasificación macroscópica e histológica (Tabla I), que dividía estos tumores en 4 grados de malignidad, con base al riesgo de aparición de metástasis (7).
Objetivos
Teniendo en cuenta los cambios ocurridos en el abordaje clínico de los tumores de GIST durante los últimos años, el objetivo de nuestro trabajo fue el de identificar los casos diagnosticados como GIST en la población de nuestro hospital, y clasificarlos y caracterizarlos de acuerdo con las nuevas clasificaciones existentes.
Material y métodos
Procedimos a un estudio retrospectivo de pacientes con diagnóstico de GIST, lesión submucosa o leiomioma, identificados por criterios inmunohistoquímicos, desde enero de 1997 hasta diciembre de 2007.
Los bloques utilizados eran representativos de las lesiones y fueron reexaminados y reclasificados en relación a la celularidad y al número de mitosis, por los criterios de Fletcher, en láminas teñidas con hematoxilina-eosina. Fueron excluidos pequeños leiomiomas con origen en la muscular de la mucosa.
Fue realizada una evaluación demográfica, del perfil y de la evolución, por acceso al proceso clínico o por consulta directa con el paciente.
En todos los pacientes se efectuó el estudio inmunohistoquímico para CD 117, CD 34, vimentina, actina del músculo liso y proteína S100 (Dako® o Novocasta®) realizando observación por microscopía electrónica en los casos que eran CD 117 negativos.
En 10 pacientes se realizó estudio de la mutación en la proteína KIT (exones 9, 11, 13 y 17); los criterios de selección fueron pacientes con edad muy joven (15 años) o con enfermedad agresiva (con metástasis o de alto riesgo, de acuerdo con los criterios de Fletcher).
No fue realizado estudio de la mutación PDGFRA.
Resultados
Se incluyeron 43 pacientes (24 hombres y 19 mujeres) con edad media de 62,7 años, desde 15 hasta 86 años cuyas características están descritas en la tabla II.
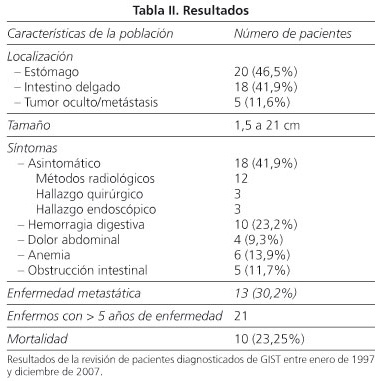
Todos los tumores fueron evaluados macroscópicamente como piezas quirúrgicas totalmente resecadas, a excepción de uno cuyo diagnóstico inicial fue por observación de biopsia endoscópica y resecado posteriormente. Nueve piezas quirúrgicas tenían invasión de los márgenes.
En la evaluación inmunohistoquímica se obtuvo positividad en 37 tumores (86%) para CD 117, 22 (51,2%) para CD 34, 12 (27,9%) para vimentina, 7 (16,3%) para actina y 1 (2,3%) para desmina; ninguno fue positivo para proteína S100. Los tumores que no expresaron proteína KIT (CD 117) fueron evaluados por otros parámetros inmunohistoquímicos y por microscopía electrónica antes de ser clasificados como GIST.
Por los criterios de Fletcher 19 casos (44,2%) eran de alto riesgo, 7 (16,3%) de riesgo intermedio, 12 (28%) de bajo riesgo y 5 (11,6%) de riesgo indeterminado, pues fue identificada la metástasis y no el tumor original. Atendiendo a la existencia de metástasis consideramos un total de 24 casos de alto riesgo (55,8%).
Fue realizada microscopía electrónica en 8 casos, incluyendo los casos CD 117 negativos; en estos, uno de los casos reveló diferenciación muscular lisa, teniendo los restantes casos aspectos inespecíficos en relación a su diferenciación (aspectos similares a los otros 5 casos CD 117 positivos).
En los 10 pacientes en los que fue estudiada la mutación en la proteína c-KIT en 5 se encontró mutación en el exón 11, en 1 fue no concluyente para el exón 9 y normal para los demás exones, en otro fue no concluyente para todos los exones y 3 no presentaban mutación. Los casos no concluyentes se deben a la baja calidad del ADN obtenido, que no permitió el análisis mutacional; la causa más probable de baja calidad del ADN se relaciona con el proceso de fijación tumoral.
Hubo 10 fallecimientos (tasa de mortalidad global: 23,25%) asociados a progresión de la enfermedad, todos pacientes con criterios de alto riesgo o indeterminado. Tan sólo una de las muertes tenía menos de 5 años de enfermedad; 9 pacientes tenían más de 5 años de enfermedad, por lo que la tasa de mortalidad englobando estos pacientes es del 42,86%. Hubo metástasis en 13 pacientes (30,2%), 5 de los cuales ya han fallecido.
Los pacientes con menos de 5 años de enfermedad (22 pacientes) aún no tienen suficiente tiempo de seguimiento pero 5 ya tienen enfermedad diseminada y 1 ya ha fallecido.
Discusión
Diagnosticamos 43 casos de GIST durante un periodo de tiempo de 10 años. La incidencia de los GIST es de 10 a 20 casos por millón de habitantes y año, habitualmente en pacientes con más de 50 años (4,6). En nuestro caso encontramos una incidencia media de 4,3 caos por 600.000 habitantes.
En nuestra serie, los resultados sobre edad coinciden con las publicaciones más recientes, con pocos casos por debajo de los 50 años (6, 8-12). Por otro lado, el predominio del sexo masculino no es consensual (9,12,13).
Desde el punto de vista epidemiológico, los GIST esporádicos son con mucho los más frecuentes, estando, sin embargo, descritos síndromes de GIST familiares. Los GIST familiares incluyen formas autosómicas dominantes, de penetrancia variable de los genes de las formas esporádicas; GIST asociados a neurofibromatosis tipo 1; y GIST asociados a paragangliomas, por pérdida alélica en genes de las subunidades de la succinito-deshidrogenasa (díada Carney-Stratakis) (13,14). No encontramos ningún caso familiar en nuestra serie.
Los GIST aparecen en todo el tubo digestivo, más frecuentemente en el estómago (60-70%), también en intestino delgado (20-25%), colon y recto (5%) y esófago (< 5%), o en localizaciones extraintestinales como mesenterio o epiplón adyacente (4,13). En nuestro caso, así como en otras casuísticas (9,10,13), encontramos apenas una pequeña diferencia porcentual entre la localización gástrica y la del intestino delgado y no encontramos ninguna localización extraintestinal. Otras revisiones recientes no encuentran diferencias entre la localización gástrica o del intestino delgado (8) o incluso verifican predominio del intestino delgado (6) (Tabla III).
La forma de presentación clínica es muy variable, pudiendo aparecer como pequeños nódulos asintomáticos encontrados accidentalmente junto con tumores agresivos (1,4) y cuya sintomatología es inespecífica (dolor o molestia abdominal, masa palpable, hemorragia digestiva) (2). La variabilidad de presentación clínica está representada en la literatura, variando desde un gran número de hallazgos accidentales (6), como en nuestro caso, hasta algunas series con prácticamente sintomatología en todos los pacientes (8,10-12).
Existen algunas publicaciones en las que se describen tumores detectados en el estómago y recto pequeños y con menor potencial maligno, al contrario de los del colon y esófago que se presentan como neoplasias avanzadas de alto riesgo (1,4). Nuestros resultados no pueden corroborar estos datos ya que no detectamos GIST de esófago, colon y recto, y encontramos lesiones de alto riesgo en el estómago. Sin embargo, en 5 pacientes apenas fueron encontradas metástasis y no el tumor primario, lo que también ya fue referido por otros autores (6,13).
Como tienen origen en el estroma, crecen generalmente como masas endo- o exofíticas bien definidas. Debido a su variabilidad clínica no existe un protocolo estipulado de diagnóstico, por lo que todas las técnicas de imagen o endoscópicas pueden ser utilizadas para su detección y localización. Así, endoscópicamente se comportan como lesiones submucosas cubiertas por mucosa normal o ulcerada (1) y radiológicamente como lesiones extrínsecas del TD (1).
El principal método diagnóstico es la ecoendoscopia, pero el diagnóstico definitivo sólo es posible con biopsia de aspiración asociada (2). La tomografía axial computerizada (TC) es muy útil para la evaluación del tamaño real, extensión local y estadificación sistémica.
La tomografía por emisión de positrones (PET/CT-FDG) corporal es también un buen método de estadificación y constituye el método más objetivo de evaluación de respuesta a la terapéutica (con los actuales inhibidores de tirosín-quinasa) en la evaluación de las situaciones marginalmente resecables en TC. Sin embargo una correcta evaluación de variación de densidades en tomodensitometría (unidades HU) constituye una alternativa en casos en que no hay posibilidad de PET.
El principal desafío consiste en diferenciar los GIST de otros tumores mesenquimatosos de células musculares lisas, como leiomioma o leiomiosarcoma, de tumores neurogénicos, como schawnoma o neurofibrosarcoma o otros tumores (como lipoma, liposarcoma, tumor carcinoide, fibroma, tumor desmoide, polipo inflamatorio fibroide) (3,4).
Como el comportamiento clínico de los GIST es variable, son todos considerados potencialmente malignos. Así, dejaron de ser clasificados en benigno o maligno, se abandonó el concepto de potencial indeterminado y se intentó encontrar clasificaciones que estratifiquen las lesiones de acuerdo con el riesgo de aparición de metástasis (5).
De acuerdo con los criterios de Fletcher, los tumores con bajo índice mitótico (≤ 5 mitosis) o de menor tamaño (< 2 cm) tienen generalmente un comportamiento poco agresivo. Sin embargo, a lo largo de los últimos años, después de una evaluación de largos seguimientos en pacientes con GIST, se ha concluido que cerca del 50% de los GIST resecados recurren en 5 años y que más del 50% de los tumores de alto riesgo recurren o desarrollan metástasis en 10 años. Además, han aumentado las descripciones de tumores mitóticamente inactivos que diseminan para otras localizaciones (4,5). Nuestros resultados están de acuerdo con la literatura, con los pacientes clasificados con criterios de bajo riesgo teniendo enfermedad menos agresiva. Confirmamos también la agresividad de estos tumores, con apenas la mitad de las muertes apareciendo en enfermedad diseminada (5 en 10 pacientes) y verificándose 30,2% de metástasis, en las que en 5 casos fueron estas las detectadas y no el tumor de origen.
El local de origen y las características radiológicas también pueden ser factores de pronóstico, aunque no tan sensibles o rigurosas (5). Radiológicamente los tumores con más de 5 cm, lobulados, heterogéneos, con infiltración de la grasa mesentérica, con linfadenopatías regionales o crecimiento exofítico, observados en TC, tienen más probabilidad de desarrollar metástasis (5). Otros autores refieren que una vascularización rica es sinónimo de mayor agresividad del tumor (8,16).
El tratamiento definitivo, después de ecoendoscopia, TC y/o PET/CT-FDG de estadificación para exclusión de metástasis, es la resección quirúrgica, con remoción completa del tumor, manteniendo intacta la cápsula y con márgenes negativos (2,17). No es necesaria la linfadenectomía ya que los GIST no diseminan para los ganglios linfáticos, pero sí para el peritoneo y el hígado, siendo por eso necesaria una exploración minuciosa intra-abdominal (2,8,17). El pronóstico depende del tamaño del tumor y del tipo de resección. Una resección completa tiene una supervivencia a los 5 años del 34 al 60% (7), hecho que nosotros también confirmamos.
La quimioterapia o radioterapia no son eficaces. La terapéutica farmacológica se basa en el uso de inhibidores (no selectivos) de la tirosín-quinasa: imatinib y sunitinib. El uso del imatinib se extiende a la enfermedad con metástasis o residual, con intención neoadyuvante y habiendo datos positivos en el contexto adyuvante. El sunitinib es usado como segunda línea, cuando fracasa el imatinib (después de haber aumentado la dosis) o en los casos de intolerancia al imatinib (15).
Las mutaciones de la proteína c-kit que incluyen el exón 11 tienen peor pronóstico (17). No podemos discutir esta afirmación ya que en nuestra serie sólo fue estudiada en pacientes con enfermedad más agresiva.
Conclusiones
El gran problema de estos tumores considerados malignos es su dificultad diagnóstica y pronóstica, debido a su escasa e inespecífica sintomatología, donde se destacan 41,9% de diagnósticos accidentales.
Los casos observados se encuentran de acuerdo con la literatura, con predominio del sexo masculino, en mayores de 50 años, asintomáticos y con mayor frecuencia de localización gástrica.
La mayoría de los casos tiene mal pronóstico, ya sea por los criterios de Fletcher, ya sea por la presencia de metástasis. Se encontró mutación de la proteína KIT en 5 de los 10 pacientes analizados, valor inferior al de la literatura. La mortalidad observada (23,25%) fue menor que la esperada ya que 22 pacientes tienen seguimiento inferior a 5 años. La mayoría de las muertes fueron en pacientes con más de 5 años de enfermedad por lo que la supervivencia de 50% a los 5 años descrita anteriormente es similar a la observada. La aplicación de los criterios de Fletcher fue consistente con la evolución, revelándose un buen método pronóstico. Se destaca la importancia de valorar las lesiones submucosas detectadas muchas veces casualmente, ya que la mayoría serán GIST, y tienen potencial de aparición de metástasis y alta tasa de recidiva y mortalidad.
Bibliografía
1. Darnell A, Dalmau E, Pericay C, Musulén E, Martín J, Puig J, et al. Gastrointestinal stromal tumors. Abdom Imaging 2006; 31(4): 387-99. [ Links ]
2. Shinomura Y, Kinoshita K, Tsutsui S, Hirota S. Pathophysiology, diagnosis and treatment of gastrointestinal stromal tumors. J Gastroenterol 2005; 40(8): 775-80. [ Links ]
3. Kuhlgatz J, Sander B, Golas M, Gunawan B, Schulze T, Schulten HJ, et al. Differential diagnosis of gastrointestinal leiomyoma versus gastrointestinal stromal tumor. Int J Colorectal Dis 2006; 21(1): 84-8. [ Links ]
4. Miettinen M, Lasota J. Gastrointestinal stromal tumors - definition, clinical, histological, immunohistochemical and molecular genetic features and differential diagnosis. Virchow Arch 2001; 438: 1-12. [ Links ]
5. Demetri G, Morgan J. Gastrointestinal stromal tumors, leiomyomas and leiomyosarcoma of gastrointestinal tract. Available at: www.uptodate.com; April 2006. [ Links ]
6. Salazar L, Gago T, Rubiales A, Jiménez B, de la Fuente R, Hernández M. Gastrointestinal stromal tumors: clinical aspects. Rev Esp Enferm Dig 2007; 99(1): 19-24. [ Links ]
7. Fletcher CD, Berman JJ, Coreless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol 2002; 33: 459-65. [ Links ]
8. Hidalgo J, Casares F, Pena S, Torres A, Bru C, Delgado J, et al. Gastrointestinal stromal tumors: factors predictive of survival after R0-cytoreduction. Rev Esp Enferm Dig 2007; 99(12): 703-8. [ Links ]
9. Rubió J, Marcos-Gragera R, Ortiz MR, Miró J, Vilardell L, Gironès J, et al. Population-based incidence and survival of gastrointestinal stromal tumours in Girona, Spain. Eur J Cancer 2007; 43: 144-8. [ Links ]
10. Bertolini V, Chiaravalli AM, Klersy C, Placidi C, Marchet S, Boni L, et al. Gastrointestinal stromal tumors - frequency, malignancy and new prognostic factors: the experience of a single institution. Pathol Res Pract 2008; 204(4): 219-33. [ Links ]
11. Hassan I, You Y, Shyyan R, Dozois E, Smyrk T, Okuno S, et al. Surgically managed gastrointestinal stromal tumors: a comparative and prognostic analysis. Ann Surg Oncol 2008; 15(1): 52-9. [ Links ]
12. Ahmed I, Welch NT, Parsons SL. Gastrointestinal stromal tumors - 17 years experience from Mid Trent Region (United Kingdom). Eur J Surg Oncol 2008; 34(4): 445-9. [ Links ]
13. Alvarado-Cabrero I, Vázquez G, Santiesban S, Hernández-Hernández DM, Pompa AZ. Clinicopathologic study of 275 cases of gastrointestinal stromal tumors: the experience at 3 large medical centers in Mexico. Ann Diagn Pathol 2007; 11(1): 39-45. [ Links ]
14. Stratakis C. Familial Gastrointestinal stromal tumors and germ-line mutations. N Eng J Med 2007; 357(10): 1054-6. [ Links ]
15. Gupta P, Tewari M, Shukla H. Gastrointestinal stromal tumors. Surg Oncol 2008; 17(2): 129-38. [ Links ]
16. Fukuta N, Kitano M, Maekawa K, Chikugo T, Kudo M. Estimation of malignant potential of gastrointestinal stromal tumors: the value of contrast-estimated coded phase-inversion harmonics US. J Gastroenterol 2005; 40(3): 247-55. [ Links ]
17. Hinz S, Pauser U, Egberts JH, Schajmayer C, Tepel J, Fandrich F. Audit of a series of 40 gastrointestinal stromal tumour cases. Eur J Surg Oncol 2006; 32(10): 1125-9. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Paula Sánchez Otero.
Hospital Fernando Fonseca, 1 C - 19.
Amadora 2720-276. Lisboa, Portugal.
e-mail: paulsanchezotero@hotmail.com
Recibido: 23-06-08.
Aceptado: 29-09-08.

