










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.101 no.3 Madrid mar. 2009
Neuroendocrine tumors - fascination and infrequency
Tumores neuroendocrinos: fascinación e infrecuencia
M. J. Varas Lorenzo
Centro Médico Teknon. Barcelona, Spain
ABSTRACT
In this article, I review and update of gastro-entero-pancreatic neuroendocrine tumors, which so much fascination have risen among healthcare providers on grounds of their infrequency, hormonal syndromes, and high survival rate, is performed based on references from the past fifteen years.
Key words: Neuroendocrine tumors. Neuroendocrine pancreatic tumors. Gastro-entero-pancreatic endocrine tumors. Carcinoid tumors.
RESUMEN
Se efectúa una revisión y puesta al día, basándose en citas bibliográficas de los últimos quince años, de los tumores neuroendocrinos gastroenteropancreáticos, que tanta fascinación han provocado en el estamento médico por su infrecuencia, síndromes hormonales y supervivencia elevada.
Palabras clave: Tumores neuroendocrinos. Tumores neuroendocrinos pancreáticos. Tumores endocrinos gastroenteropancreáticos. Carcinoides.
Definition
Neuroendocrine tumors (NETs) or gastroenteropancreatic endocrine tumors (GEPETs) (2% of all gastrointestinal tumors) have their origin in tissues derived from the neural crest, neuroectoderm, and endoderm; 60-70% are carcinoid tumors, and 20-40% are found in the pancreas (NEPTs) (Tables I and II). Gastrointestinal tract endocrine tumors other than carcinoid tumors may be gastrinomas, enteroglucagonomas, somatostatinomas, or non-functioning neuroendocrine tumors. Many of the data discussed herein have been taken from prior reviews (1-5).
Incidence
In necropsies 0.5-1.5%, or even up to 5%, are NEPTs (1-2), many of them silent. They represent 1-3% of all pancreatic neoplasms.
In Europe, in the one community-based study carried out thus far, incidence is smaller than one case per 100,000/year - specifically, 0.32 per 100,000/year for carcinoid tumors and 0.2 per 100,000/year (2 per million/year) in the pancreas (6). Insulinoma and gastrinoma, the most common types, have an incidence of 1 new case per million people and year. Glucagonoma and somatostatinoma are least common (1 per 20 and 40 million/year, respectively) (Table II).
Carcinoids are twice as common in Afroamerican patients than among Caucasians (7).
In the USA (8), incidence approaches 5 per million/year for NEPT, 1.8 in women and 2.6 in men, but is increasing and may well reach 10 per million people/year, with a higher frequency between 40 and 60 years of age, and in women.
In our country the incidence of NEPT may be 0.08 per 100,000 inhabitants/year, or 1 new case every two years in Hospitalet de Llobregat (Barcelona).
Genetics
Tumors may be sporadic, particularly carcinoids, or may develop in association with dominant autosomic syndromes (2) - locus 11q13 for MEN-1 - including multiple endocrine neoplasm (MEN; types 1 and 2; a and b), von Hippel-Lindau disease (VHL), von Recklinghausen's neurofibromatosis, and tuberous sclerosis (5) (Table III).
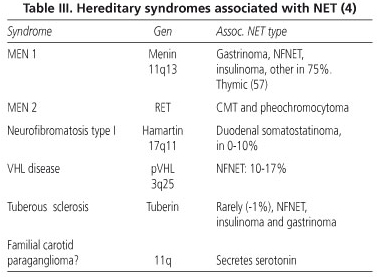
MEN-2 results from a mutation in the RET proto-oncogene.
The risk for carcinoid development is also accounted for by genetic factors. Indeed, gastric NETs are increased in women with diabetes and a family history of cancer (9).
Functioning and non-functioning tumors (NFNET)
NFNETs are diagnosed during routine ultrasonography (Fig. 1), CT scans for unexplained abdominal symptoms, or late-stage disease (70% greater than 5 cm).

Their frequency is 20-58%, and up to 90% when located in the pancreas (10-12).
Most (over 70%) are not really functioning, since they secrete substances such as PP, other peptides (PYY, ghrelin, etc.), NSE, CgA, or HCG, but none of these substances results in specific symptoms.
Functioning or secretory tumors (60%) may result in multiple hormone secretion (MHS) or be associated with MEN, with well-defined syndromes.
Except for insulinoma (less than 10%) many NETs are malignant (50-60% or higher, particularly NFNEPT) and result in nodal, hepatic (most commonly), bony (12%), pulmonary (4%), and cerebral metastases (Table II).
Overall survival at 5 years never exceeds 67% when all NETs are considered. WHO has recently issued a classification system for NETs: well-differentiated tumor, well-differentiated carcinoma, and poorly-differentiated carcinoma, according to histology, size, and proliferation indices; a TNM (tumor, node, metastasis) classification had also been proposed earlier (5).
Clinical suspicion
NEPTs that inappropriately secrete peptides and hormones (FNEPTs) result in specific clinical syndromes with guide symptoms (13-15). Thus, the development of acute and/or chronic hypoglycemia (guide symptom) should prompt suspicion for hyperinsulinism due to pancreatic insulinoma (Table II).
The emergence of abdominal pain, vomiting and diarrhea in a patient with ulcer, particularly when Helicobacter pylori-negative, should alert on the presence of Zollinger-Ellison syndrome (ZES) due to pancreatic or extrapancreatic gastrinoma (14).
Glucagonoma syndrome is characterized by necrolytic migratory erythema (NME) (52%) and diabetes (22%) (16); somatostatinoma and corticotropinoma are also accompanied by hyperglycemia.
Verner-Morrison syndrome (VMS) from vipoma presents with secretory diarrhea and hypopotasemia (70-100%). Many NEPTs produce chronic diarrhea, but only vipoma and neurotensinoma lead to hypokalemic diarrhea.
The clinical "inhibitory" somatostatinoma syndrome combines diabetes with bladder disease, diarrhea and hypochlorhydria. Somatostatinoma may arise in the duodenum or pancreas, with malignity in excess of 43% (17).
Flushing is a typical finding of carcinoid syndrome (CS) that develops in 10% of carcinoid tumors (Table IV).

Clinical suspicion should lead physicians to order a hormone panel (MHS and MEN should also be ruled out), but on occasion tumors are non-functioning or hormone levels are within the normal range, and recourse to hormone provocation tests is required (14,15,18).
Biochemical diagnosis
Pancreatic polypeptide (PP), neuron-specific enolase (NSE), chromogranins (A, B, C), synaptophysin (P38), 7B2, and HCG are nonspecific markers with special relevance for the diagnosis of NFNEPTs.
CgA and NSE are excellent markers for GEPET, whereas Ki 67 (higher than 2%) or MIB-1 is a malignity predictor (Table V). CgA is probably more specific and NSE, more sensitive (83-100%).

CgA has a sensitivity of 60-100% in patients with metastatic disease, but lower than 50% in patients with local tumors (15).
Among FNEPTs and carcinoids insulin, glucagon, somatostatin, gastrin, VIP, PP, serotonin (and its metabolite 5-HIAA in urine), etc., are titrated by RIA.
This hormone panel using RIA is relevant not only for biochemical diagnosis but also for follow-up, for the monitoring of response to chemotherapy and surgery (14,15).
Measuring PTH, calcium, calcitonin, prolactin, etc., is also mandatory.
Diagnosis and imaging techniques
Gut NETs (mainly carcinoids) are localized using radiography and endoscopy (Fig. 2), and may be staged by endoscopic ultrasonography (EUS), which provides information about the depth and extension of the tumor, and that in turn facilitate endoscopic tumorectomy or polypectomy (1-2-cm carcinoids in superficial layers with no muscularis propria infiltration or adenopathies). Metastatic disease and occult tumors should be excluded with octreoscan and PET.

NEPTs are more difficult to localize; almost 20-30% of cases fail to be localized with imaging techniques (ultrasounds, HCT, gadolinium MRI, scintigraphy, octreoscan, etc.).
CT and MRI detect 30-94% of cases, and octreoscan detects 80-90% (except insulinoma: 50%), while EUS detects 79-100% (5).
Effectively, pancreatic EUS visualizes tumors as small as 3 mm with a sensitivity above 85% when located intrapancreatically in the head or body; even, in a study that analyzed a larger sample the sensibility was 93% in a paper published reporting a greater sample (19).
Literature reviews (20-22) suggest a high mean sensitivity at 88% with a specificity of 85%; hence, around 5-10% would still remain hidden preoperatively.
In our cumulative experience with 50 patients, 35 of whom had 34 tumors operated (14 in the pancreas, 20 in the gut, 15 carcinoids), EUS precision was 82.5%, with a sensitivity of 88% (94% for carcinoids, 90% for gut tumors, 86% for pancreatic tumors) and a PPV of 91%. Three tumors smaller than 10 mm, 4 smaller than 15 mm (all insulinomas), and 1 cystic tumor 4 mm in size (2%), which was seen to be a functioning gastrinoma, were detected in the pancreas.
Outcomes over 10 years suggest a gradual increase in diagnostic accuracy and sensitivity as a result of experience (22).
EUS-FNAP was used for 10 cases (8 were operated upon) with an accuracy of 75%, sensitivity of and PPV of 83%, and specificity and NPV of 50%, since there was a false positive result (Table VI; Fig. 3.)
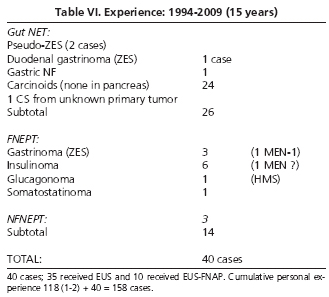

A localization and staging algorithm has been proposed based on the use of latest-generation helical CT and both radial and sectorial EUS when tissue samples are to be obtained with FNAP; accuracy and sensitivity are variously reported in the literature, but above 80% on average, and higher than 90% in the series with a greater use of FNAP (23).
In some cases (1.5-4%) (24,25), particularly very small NEPTS, most non-functioning masses (24-27), may show a cystic pattern, and EUS-FNAP may be successful in (26,27) in nearly 100% of cases.
Prevalence of NEPTS is estimated to be potentially higher at 9.5% (26), but our experience is similar (2%) to that reported in the reviewed literature (25).
Currently, the best strategy is likely the use of HCT plus optional EUS-FNAP. Octreoscan may be replaced by octrotide PET in the future, with a sensitivity, specificity and accuracy above 90%.
These results may change in the future with the use of novel technologies such as USI, color EUS or EUS with Sonovue contrast (56), sonoelastography, intraductal ultrasonography using miniprobes, PET, etc.
Therapeutic options
1. Surgery. Classic or laparoscopic surgical therapy with total excision presently represents the only curative alternative after patient stabilization (fluids, diazoxide, PPIs, insulin... depending on tumor type).
Endoscopic polypectomy is indicated for digestive tract carcinoids smaller than 1-2 cm, located in superficial layers with no muscularis propria involvement, and in the absence of adenopathies (22).
2. Somatostatin and somatostatin analogs (octreotide, lanreotide, pasireotide). They inhibit hormone secretion. They may be administered via the subcutaneous or intramuscular route daily or every 7-14-30 days at doses oscillating between 100 and 1500 mcg/day in two to three subcutaneous injections. For intramuscular use 20-30 mg are administered every 7-14-28 days based on response - even 20 mg monthly. They do not regress tumors (3-14%) but do stabilize 36-70% of masses, and reduce clinical (> 50%) and biochemical (43%) parameters (28).
3. Interferons. Human leukocytic interferon and human recombinant interferon alfa-2B are administered subcutaneously, the latter at a dosis of 2 to 6 million IU/day for several weeks, for a tumor response of 11% and a biochemical response of 44% on average (29,30). According to a recent study with pegylated interferon alfa the disease would stabilize in 75% of cases (31) with symptom management.
4. Chemotherapy. With streptozotocine (1 g i.v./m2/day in 5-7-day courses every 6 weeks), streptozotocine plus 5-FU, or doxorubicin; this leads to remission in up to 60% of patients. Combined cisplatin and etoposide may be used for poorly-differentiated NETs.
Oral temozolomide and thalidomide are used in patients with advanced metastatic disease (32).
5. Radiotherapy and other treatments. Palliative radiotherapy with In111-DTPA octreotide for non-resectable tumors with a positive octreoscan test, with partial remission and tumor size stabilization in over 60% of cases.
NETs may release multiple growth factors, vascular endothelial growth factor, platelet-derived growth factor, insulin-like growth factor, fibroblast growth factor, transforming growth factor, and epidermal growth factor, as well as their respective receptors. New molecules are used to target these factors and their receptors, including monoclonal antibodies, mTOR inhibitors, tyrosine kinase inhibitors such as sunitinib, with a response rate lower than 20% (5); a recent report (33) of 107 patients, how-ever, 44% of carcinoids and 62% of NEPTs exhibited at least some tumor regression and estabilization.
6. Combinations of the above. For instance, somatostatin analogs plus interferon alfa (30) or chemotherapy.
7. For hepatic metastases. Chemoembolization, ethanolization, criosurgery, and radiofrequency ablation; ultimately liver transplantation may be considered for younger patients with liver metastases and excised primary NEPT (15).
Prognosis and survival
The prognosis of carcinoids is very poor when carcinoid syndrome develops (10% of cases), as this is a manifestation of advanced disease (liver metastasis).
The prognosis of NEPTs depends on functioning status, MEN status, primary tumor size and site, and presence of metastatic disease (tumor extension) (14).
According to Massachussets General Hospital statistics (168 NEPTs) 76% were benign, and 26% were malignant with liver metastasis. Actuarial survival was 77 and 62% at 5 and 10 years (10).
In benign NEPTs 5-year survival was 92, vs. 50% for malignant NEPTs (11).
Today, the primary concern of specialists regarding ZES is the development of liver metastases, which represent the most significant factor for poor prognosis.
When operated on with curative intent, gastrinoma shows a 15-year survival rate of 98%, versus 74% for non-surgical patients (34). In all, 29% of non-operated patients developed liver metastases.
According to Princess Margaret Hospital statistics (193 NET in 10 years: 72% carcinoids and 21% NEPTs) 5-year survival was 58%; in the multivariate analysis, age, primary site, and surgery with curative intent were independent predictors of survival. When curative surgery was attempted, survival at 5 years increased to 86% (35).
According to Mayo Clinic statistics (1483 NETs over 27 years with a high percentage of non-functioning tumors: 91%) advanced status and age were predictive for poorer survival, and better results were obtained for functioning versus non-functioning growths (8).
Patients operated on for MEN-1 have similar survival at 7-10-15 years, but with high recurrence rates; only 4.5% are free from tumors at 10 years (36).
Survival for NET without liver metastasis was 95-90-83% at 5-10-15 years, respectively; even for malignancy, survival at 5 years may be up to 77-95% when aggressive treatment is attempted with primary tumor resection and adjuvant therapy, versus 36% at 5 years in other studies (4).
Several aspects are associated with poor prognosis: tumor size greater than 2 cm, presence of vascular or perineural invasion, pancreatic capsule infiltration, histological differentiation (poor differentiation), mitosis numbers, cell atypia, high Ki67 (higher than 30%), and presence of liver or lymphatic metastases.
In 70 patients operated upon for NEPT (23% insulinomas, 71% non-functioning, 53% malignant) actuarial survival at 5 years was 77%. Positive nodes, perineural invasion, and liver metastasis had no impact on survival, only lympho-vascular invasion had it (37).
German authors (38,39) having studied nearly 400 NETs, 70% with metastasis at diagnosis, believe that tumor size smaller than 2.5 cm, surgery, absence of metastasis and symptoms, and low CgA and Ki67 are best as survival markers.
The Swedish team (40) studied prognostic factors in 324 patients with NEPT, with survival rates at 5 and 10 years of 64 and 44%, respectively. In the univariate analysis, tumor stage, radical surgery, non-functioning status, high Ki67 and CgA, tumor size, and sporadic (non-familial) nature appeared as significant prognostic factors; in the multivariate analysis only stage, radical surgery, and Ki 67 > 2% remained so.
Italian authors (41) studied 180 cases of NFNEPT with survival at 5, 10, and 15 years of 67, 49, and 33%, respectively, and confirmed that metastasis, poor differentiation, Ki67, and weight loss are prognostic factors (Table V).
American authors (42) studied a registry with 35,825 cases, and concluded that in the multivariate analysis predictive factors included tumor differentiation, stage, primary tumor location, histological grade, sex, race, age, and year of diagnosis; they confirmed increased incidence, which changed from 1 per 100,000 in 1973 to 5.25 per 100,000 in 2004.
These data are consistent with incidences (1993-2004) in the USA and Norway, 4.44 versus 3.24 in Caucasian patients, and higher in Afroamerican subjects. Most frequent were always bronchopulmonary tumors (32%). Survival at 5 years for all NETs was 55% (43).
Regarding bronchopulmonary NET, most examples are little-functioning carcinoid tumors (-5%) with Cushing syndrome, acromegaly, and SIADH. Incidence in 2003 was 1.57 per 100,000, and on the rise. Survival at 5 years was 88% for typical carcinoid tumors with scarce growth (44).
GEPETs were previously considered to be more common than bronchopulmonary tumors, albeit a recent registry of 1,000 cases in Brazil showed 72% in the thorax, 20% GEPETs, and 3.6% in unknown places (45).
Summary and conclusions
Neuroendocrine tumors (NETs) or gastro-entero-pancreatic endocrine tumors (GEPETs) may be carcinoids in 70% and pancreatic in 20% of cases.
Their incidence is higher than 5 cases per million population per year, and is currently increasing.
They may be sporadic or linked to genetic mutations.
They may be functioning (50%) or non-functioning; benign or malignant as a function of the presence of metastatic disease (50-60%).
Clinical suspicion should lead physicians to order a hormone panel with nonspecific markers such as CgA and NSE, or specific markers including insulin, gastrin, VIP, serotonin -5-HIAA, etc.
Currently, the diagnostic algorithm for gut carcinoids includes endoscopia with biopsy, HCT, and octreoscan vs. PET.
For NEPT and MEN, the best strategy is seemingly HCT plus radial or sectorial EUS with optional FNAP (Fig. 3), followed by octreoscan vs. PET.
PET using 68Ga-DOTA-Tyr3-octreotide has a sensitivity of 97%.
Following patient stabilization for inappropriate hormone secretion (fluids, diazoxide, PPIs, insulin, etc.), in the absence of metastasis surgery should be attempted with curative intent; when metastasis is present, adjuvant therapy should be added.
In advanced disease, treatment should be attempted with somatostatin analogs, interferons, both, or even chemotherapy or radiation therapy with octreotide when a positive octreoscan is obtained.
Prognosis and survival mainly depend on potential curative-intent surgery, the absence of advanced metastatic disease, and probably lymphovascular invasion (37).
Additional significant factors include primary pancreatic tumor size above 2.5 cm, high CgA and Ki67 (38-40), and metastasis, Ki67, and weight loss for NFNEPT (41).
For gut carcinoids (Fig. 4) smaller than 1 cm, endoscopic polypectomy is satisfactory and has replaced classic surgery for tumors not infiltrating the muscularis propria and with no metastatic disease (46). In such cases, and in appendicular carcinoids, 5-year survival rates are above 95%, while they decrease to 25 and 65% in the presence of liver or nodal metastases (47-49).
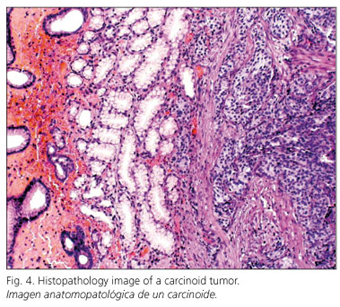
Despite recent advances in the diagnosis and treatment of NEPT (48,49), survival improvement remains unclear. In a recent review (51) no changes have been recorded for survival rates during the past 30 years regarding all GEPETs (carcinoids and NEPTs) (52).
Recent papers advocate for the surgical resection of pancreatic tumors (53), but in patients with metastatic disease primary tumor resection is not beneficial for midgut-derived carcinoids (54).
Four cases of GEPET have been recently reported in patients with HIV infection (55).
References
1. Varas MJ. Endocrinología Gastroentero-pancreática. Madrid: Smar SL.; 1997. [ Links ]
2. Varas MJ. Tumores de los islotes pancreáticos. En: Vilardell F, et al., editores. Enfermedades Digestivas 2. Madrid-Barcelona: Aula Médica; 1998. p. 1516-24. [ Links ]
3. Fritscher-Raven A. Endoscopic ultrasound and neuroendocrine tumours of the pancreas. JOP 2004; 5(4): 273-81. [ Links ]
4. Mougey AM, Adler DG. Neuroendocrine tumors: review and clinical update. Hospital Physician 2007. p. 12-20. [ Links ]
5. Massironi S, Sciola V, Peracchi M, Ciafardini C, Spampatti MP, Conte D. Neuroendocrine tumors of the gastro-entero-pancreatic system. World J Gastroenterol 2008; 14(35): 5377-84. [ Links ]
6. Buchanan KD, Johnston CF, O'Hare M, Ardill JE, Shaw C, Collins JS, et al. Neuroendocrine tumors: a European view. Am J Med 1986; 81: 14-27. [ Links ]
7. Kang H, O'Connell JB, Leonardi MJ, Maggard MA, MxGory ML, Ko CY. Rare tumors of the colon and rectum: a national review. Int J Colorectal Dis 2007; 22: 183-9. [ Links ]
8. Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol 2008; 19(10): 1727-33. [ Links ]
9. Hassan MM, Phan A, Li D, Dagohoy CG, Leary C, Yao JC. Risk factors associated with neuroendocrine tumors: A U.S.-based case-control study. Intern J Cancer 2008; 123(4): 867-73. [ Links ]
10. Vagefi PA, Razo O, Deshpande V, et al. Evolving patterns in the detection of pancreatic neuroendocrine tumors (PNETS): The Masschusetts General Hospital experience. Pancreas 2006; 33(4): 504 A. [ Links ]
11. Liu H, Zhang SZ, Wu YL, Fang HQ, Li JT, Sheng HW, et al. Diagnosis and surgical treatment of pancreatic endocrine tumors in 36 patients: a single-center report. Chin Med J 2007; 120(17): 1487-90. [ Links ]
12. Jagad RB, Koshariya M, Kawamoto J, Papastratis P, Kefalourous H, Patis V, et al. Pancreatic neuroendocrine tumors: our approach. Hepato-gastroenterology 2008; 55: 275-81. [ Links ]
13. Varas MJ. Síndrome de Zollinger-Ellison. Rev Med Univ Navarra 1995; 39: 18-23. [ Links ]
14. Varas MJ. Tumores endocrinos pancreáticos, ¿cuándo sospecharlos, cómo diagnosticarlos y cómo tratarlos? Revisiones en Gastroenterología 2000; 2(1): 47-55. [ Links ]
15. Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology 2008; 135: 1469-92. [ Links ]
16. Kindmark H, Sundin A, Granberg D, Dunder K, Skogseid B, Janson ET, et al. Endocrine pancreatic tumors with glucagon hypersecretion: a retrospective study of 23 cases during 20 years. Med Oncol 2007; 24(3): 330-7. [ Links ]
17. Garbrecht N, Anlauf M, Schmitt A, Henopp T, Sipos B, Raffel A, et al. Somatostatin-producing neuroendocrine tumors of the duodenum and pancreas: incidence, types, biological behavior, association with inherited syndromes, and functional activity. Edocr Relat Cancer 2008; 15(1): 229-41. [ Links ]
18. Berna MJ, Hoffmann K, Long SH, Serrano J, Gibril F, Jensen RT. Serum gastrin in ZES: II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature. Evaluation of diagnostic critera, proposal of new criteria, and correlations with clinical and tumoral features. Medicine 2006; 85(6): 331-64. [ Links ]
19. Anderson MA, Carpenter S, Thompson NW, Nostrant TT, Elta GH, Scheiman JM. Endoscopic ultrasound is highly accurate ad directs managements in patients with neuroendocrine tumors of the pancreas. Am J Gastroenterol 2000; 95(9): 2271-7. [ Links ]
20. Varas MJ, Armengol JR, Boix J, et al. Diagnóstico y localización preoperatoria de los tumores endocrinos digestivos mediante ultrasonografía endoscópica. Gastroenterol y Hepatol 1999; 22: 223-6. [ Links ]
21. Varas MJ, Miquel JM, Maluenda MD, Boix J, Armengol JR. Preoperative detection of gastrointestinal neuroendocrine tumors using endoscopic ultrasonography. Rev Esp Enferm Dig 2006; 98: 828-36. [ Links ]
22. Varas MJ. Ultrasonografía endoscópica, aplicaciones diagnósticas y terapéuticas. Madrid: Ed. Médica Panamericana; 2008. [ Links ]
23. Santo E, Kariv R, Monges G, et al. The role of linear array endoscopic ultrasound with fine-needle aspiration in the diagnosis and preoperative evaluation of pancreatic neuroendocrine tumors -experience with 76 cases. Gastrointest Endosc 2002; 56 (4): S118. [ Links ]
24. Li Destri G, Regio E, Veroux M, Lanzafame S, Puleo S, Minutolo V. A rare cystic non-functioning neuroendocrine pancreatic tumor with an unusual presentation. Tumori 2006; 92: 260-3. [ Links ]
25. Ardengh SA, Komorowski RA, Demeure MJ, Wilson SD, Pitt HA. Cystic pancreatic neuroendocrine tumors: is preoperative diagnosis possible? J Gastrointest Surg 2002; 6: 66-74. [ Links ]
26. Baker MS, Knuth JL, DeWitt J, LeBlanc J, Cramer H, Howard TJ, Schmidt CM, et al. Pancreatic cystic neuroendocrine tumors: preoperative diagnosis with endoscopic ultrasound and fine-needle immunocytology. Gastrointest Surg 2008; 12(3): 450-6. [ Links ]
27. Kongkam P, Al-Haddad M, Attasaranya S, O'Neil J, Pais S, Sherman S, et al. EUS and clinical characteristics of cystic pancreatic neuroendocrine tumors. Endoscopy 2008; 40(7): 602-5. [ Links ]
28. Varas MJ, Tortosa F. Tumores endocrinos gastroenteropancreáticos y tratamiento con octreótida. Cirugía Española 1995; 58(4): 325-8. [ Links ]
29. Varas MJ, Gordillo J. Tumores pancreáticos endocrinos y terapia con interferón alfa 2b. Gastroenterol y Hepatol 1992; 15(7): 394-6. [ Links ]
30. Arnold R, Simon B, Wied M, Öberg K. Treatment of neuroendocrine GEP tumours with somatostatin analogues. Digestion 2000; 62 (Supl. 1): 84-97. [ Links ]
31. Pavel ME, Baum U, Hahn EG, Schuppan D, Lohmann T. Efficacy and tolerability of pegylated IFN-alpha in patients with neuroendocrine gastroenteropancreatic carcinomas. J Inteferferon Cytokine Res 2006; 26: 8-13. [ Links ]
32. Kulke MH, Stuart K, Enzinger PC, Ryan DP, Clark JW, Muzikansky A, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol 2006; 24: 401-6. [ Links ]
33. Kulke MH, Lenz HJ, Meropol NJ, Posey J, Ryan DP, Picus J, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol 2008; 26(20): 3403-10. [ Links ]
34. Norton JA, Fraker D, Alexander HR, Gibril F, Liewehr DJ, Venzon DJ, et al. Surgery increases survival in patients with gastrinoma. Ann Surg 2006; 244(3): 410-9. [ Links ]
35. Rothenstein J, Cleary SP, Pond GR, Dale D, Gallinger S, Moore MJ, et al. Neuroendocrine tumors of the gastrointestinal tract: A decade of experience at the Princess Margaret Hospital. Am J Clin Oncol 2008; 31(1): 64-70. [ Links ]
36. Ellison EC, Sparks J, Verducci JS, Johson JA, Muscarella P, Bllomston M, et al. 50-year appraisal of gastrinoma: recommendations for staging and treatment. J Am Coll Surg 2006; 202: 897-905. [ Links ]
37. Kazanjian KK, Reber HA, Hines OJ. Resection of pancreatic neuroendocrine tumors: results of 70 cases. Arch Surg 2006; 141: 765-9. [ Links ]
38. Böhmig M, Pape UF, Tiling N, et al. Prognostic factors in gastroenteropancreatic neuroendocrine tumors -a retrospective multivariate analysis. J Clin Oncol 2005; 23(16S): 4086. [ Links ]
39. Pape UF, Berndt U, Müller-Nordhorn J, Böhmig M, Roll S, Koch M, et al. Prognostic factors of long-term outcome in gastroenteropancreatic neuroendocrine tumours. Endocrine-Related Cancer 2008; 15(4): 1083-97. [ Links ]
40. Ekebland S, Skogseid B, Dunder K, Öberg K, Eriksson B. Prognostic factors and survival in 324 patients with pancreatic endocrine tumor treated at a single institution. Clin Cancer Research 2008; 14: 7798-03. [ Links ]
41. Bettini R, Boninsegna L, Mantovani W, Capelli P, Bassi C, Pederzoli P, et al. Prognostic factors at diagnosis and value of WHO classification in a mono-institucional series of 180 non-functioning pancreatic endocrine tumours. Ann Oncol 2008; 19(5): 903-8. [ Links ]
42. Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35.825 cases in the United States. J Clin Oncol 2008; 26(18): 3063-72. [ Links ]
43. Hauso O, Gustafsson BI, Kidd M, Waldum HL, Drozdov I, Chan AK, et al. Neuroeendocrine tumor epidemiology: contrasting Norway and North America. Cancer 2008; 113(10): 2655-64. [ Links ]
44. Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM. Bronchopulmonary neuroendocrine tumors. Cancer 2008; 113(1): 5-21. [ Links ]
45. Younes RN, on behalf of the GETNE. Neuroendocrine tumors: A registry of 1000 patients. Rev Assoc Med Bras 2008; 54(4): 305-7. [ Links ]
46. Martinez-Ares D, Souto J, Varas MJ, Espinós JC, Yáñez J, Abad R, et al. Endoscopic ultrasound-assisted endoscopic resection of carcinoid tumors of the gastrointestinal tract. Rev Esp Enferm Dig 2004; 96(12): 847-55. [ Links ]
47. Lips CJM, Lentjes EGWM, Höppener JWM. The spectrum of carcinoid tumours and carcinoid syndromes. Ann Clin Biochem 2003; 40: 612-27. [ Links ]
48. Ramaje JK, Davies AH, Ardill J, Bax N, Caplin M, Grossman A, et al.; UKNETwork for Neuroendocrine Tumours. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut 2005; 54: iv1-iv16. [ Links ]
49. de Herder WW, O'Toole D, Rindi G, et al. ENETS consensus guidelines for the diagnosis and treatment of neuroendocrine gastrointestinal tumors: part 2 -midgut ad hindgut tumor. Neuroendocrinology 2008; 87: 1-63. [ Links ]
50. Halfdanarson TR, Rubin J, Farnell MB, Grant CS, Petersen GM. Pancreatic endocrine neoplasms: epidemiology and prognosis of pancreatic endocrine tumors. Endocrine-Related Cancer 2008; 15: 409-27. [ Links ]
51. Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 2008; 9: 61-72. [ Links ]
52. Jensen RT. Endocrine neoplasms of the pancreas. In: Yamada T, et al., editors. Textbook of Gastroenterology. 5th ed. Oxford: Blackwell; 2008. [ Links ]
53. Hill JS, McPhee JT, McDade TP, Zhou Z, Sullivan ME, Whalen GF, et al. Pancreatic neuroendocrine tumors: the impact of surgical resection on survival. Cancer 2009; 115(4): 741-51. [ Links ]
54. Strosberg J, Gardner N, Kvols L. Survival and prognostic factor análisis of 146 metastatic neuroendocrine tumors of the mid-gut. Neuroendocrinology 2009. [ Links ]
55. Lito P, Pantanowitz L, Marotti J, Aboulafia DM, Campbell V, Bower M, et al. Gastroenteropancreatic neuroendocrine tumors in patients with HIV infection: a trans-atlantic series (clinical investigation). Am J Med Sciences 2009; 337(1): 1-4. [ Links ]
56. Malagò R, D'Onofrio M, Zamboni GA, et al. Contrast-enhanced sonography of nonfunctioning pancreatic neuroendocrine tumors. AJR 2009; 192: 424-30. [ Links ]
57. Goudet P, Murat A, Cardet-Bauters C, et al. Thymic neuroendocrine tumors in multiple endocrine neoplasia type 1. Comparative study on 21 cases among a series of 761 MEN 1 from GTE (Group des Tumeurs Endocrines). World J Surg 2009; 26: 891-6. [ Links ]
![]() Correspondence:
Correspondence:
M. J. Varas Lorenzo.
Centro Médico Teknon.
Marquesa de Vilallonga, 12.
08017 Barcelona, Spain.
e-mail: varas@dr.teknon.es
Received: 28-12-08.
Accepted: 08-01-09.

