










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.101 no.6 Madrid jun. 2009
The pathogenesis of primary biliary cirrhosis
Patogénesis en cirrosis biliar primaria
J. A. Solís Herruzo, P. Solís Muñoz and T. Muñoz Yagüe
Department of Digestive Medicine. Hospital Universitario 12 de Octubre. Madrid, Spain
ABSTRACT
Primary biliary cirrhosis (PBC) would develop when the immune system comes across a microorganism with proteins similar to those in the piruvate dehydrogenase complex E2 (PDC-E2), or a neoantigen resulting from a xenobiotic-modified autoantigen. This would lead to an innate immune response where TLRs would play a pivotal mediating role, which would give rise to a local microenvironment favoring an adaptive immune response. Such response would be particularly strong in individuals with selected genetic characteristics. The genetic characteristics underlying this predisposition remain unknown, but they likely entail small numbers of scarcely-active regulatory T cells. The AE2 anion exchanger, which is deficient in patients with PBC, may reduce the number and activity of regulatory T cells. NK cells are also pivotal in the preparation of an adaptive response, as they release a number of cytokines and chemokines that favor and recruit antigen-presenting cells to activate B and T cells - CD4+ Th1 and CD8+. An activation of the former would increase the production of IgM and anti-mitochondrial IgG and IgA antibodies against PDC-E2. An activation of CD8+ cells, also sensitive to PDC-2 as aberrantly expressed on the surface of BECs and SECs, would result in apoptosis for these epithelial cells, and in small bile-duct destruction. Immune response is likely inadequately suppressed because of the small numbers of scarcely-active regulatory T cells, the latter resulting from low genetic expression and activity of the AE2 transporter.
Key words: Primary biliary cirrhosis. Pathogenesis. Autoimmunity. Xenobiotics. Genetics.
Introduction
Primary biliary cirrhosis (PBC) is an inflammatory disease of unknown etiology that affects almost exclusively women; lesions selectively compromise small bile ducts, hence the disease results in chronic cholestasis. While PBC is the most widely accepted designation for this disease, it is inappropriate since the condition has several stages with no liver cirrhosis but only chronic, nonsuppurative, destructive cholangitis.
Elementary lesions in PBC concentrate in small septal and interlobular bile ducts, where biliary epithelial cells (BECs) show various degrees of degeneration. The walls of these injured ducts commonly exhibit interepithelial CD8+ and CD4+ Th1 T cells surrounded by dense infiltrates of CD4+ and CD8+ B cells, among other cell types. Over time degenerative changes in BECs become more pronounced, and these cells and their associated bile ducts eventually disappear. In this stage epithelioid-cell aggregates or non-caseating granulomas are usually found in the vicinity of or around involved bile ducts. These lesions, which are not pathognomonic of PBC, have been designated florid duct lesions or chronic nonsuppurative destructive cholangitis (1,2).
Pathogenesis of PBC
Small bile duct lesions
The mechanism through which typical PBC lesions arise is unknown; however, enough data have been collected of late to endorse that the destruction of small bile ducts is the result of an immune aggression that leads to apoptosis in BECs (3-5).
While the deep mechanims determining apoptosis in these cells remain unknown, factors such as Fas/FasL, TRAIL (TNF-related apoptosis-inducing ligand), and Bcl-2 proteins in mitochondrial walls are known to play a role in this process (4,6-9). Indeed, PBC-related BECs have Fas on their surface, and neighboring mononuclear inflammatory cells express FasL (10-12). TNFα and IFNγ may act as mediators for BEC apoptosis, as both these cytokines increase Fas expression (13,14). TRAIL, another apoptosis mediator in several liver conditions (15), is expressed by BECs and hepatocytes in PBC (16), in addition to other inflammatory cells. Its expression increases with bile acid levels (17). The exit of cytochrome c from the mitochondrial inter-membrane space into the cytoplasm plays a key role in apoptosis. This process is facilitated by protein Bax, among others, and hindered by proteins Bcl2 and BclXL, located in the outer mitochondrial membrane (4,5,12). In PBC, Bcl2 is typically undetectable in BECs (18), hence cytochrome c might easily leak out of mitochondria to start the apoptotic mechanims.
Immune response in PBC
While molecular mechanisms determining BEC apoptosis are not clearly understood, it is accepted that autoimmune response is pivotal in the pathogenesis of lesions found in PBC. A study of immunity in patients with PBC reveals changes both in humoral and cell-mediated immune responses, some of them most likely related to small bile duct damage. Major changes include the presence of anti-mitochondrial antibodies (AMA) in the blood, an aberrant expression of HLA-II molecules and piruvate dehydrogenase complex E2 (PDC-E2) molecules on the surface of the biliary epithelium, and portal and periportal infiltration with activated CD4+ and CD8+ lymphocytes sensitized to PDC-E2. Such self-reactive T cells probably play a role in BEC apoptosis via a release of granzyme/perforin or through FasL or TNFα (3,19-21).
Humoral immunity
A most typical change in PBC is high AMA titers in more than 95% of patients (22), and both anti-nuclear (30%) and anti-centromere (< 10%) antibodies may also be found. Anti-nuclear antibodies are found particularly in the absence of AMA, and anti-centromere antibodies are detected when PBC is associated with self-limited scleroderma. AMA may appear before any other manifestation occurs, and this fact predicts that the disease will eventually manifest clinically (23-25). Only a minority of patients (5%) have undetectable AMA (26); however, these patients have clinical, histological, and cell-mediated immune changes that are identical to those of the remaining 95% (27). AMA are antibodies that recognize the oxo-acid dehydrogenase complex (OADC), including the piruvate dehydrogenase complex (PDC), 2-oxoglutarate dehydrogenase complex (OGDC), branched-chain oxo-acid dehydrogenase complex (BCOADC), and subunits E1a, Elb, and E3 of PDC binding protein or protein X (28-30). All these complexes are located in the inner mitochondrial membrane and play a role in keto-acid oxidative decarboxylation and amino-acid catabolism. The main epitope in all these enzymatic complexes, be it for AMA, CD4+ cells and CD8+ cells, resides within their lipoil domains (3,31,32).
AMA likely have no pathogenetic role. A number of facts could hardly be explained should AMA be responsible for this condition. For example: a) these antibodies persist after liver transplant when no lesions are present in grafts; b) there is no relationship between AMA titers and liver damage severity; c) some patients with PBC have no AMA and exhibit lesions identical to those seen in the presence of AMA; d) AMA formation may be induced in animal models by injecting PDC-E2, and this is associated with no biochemical changes or PBC-related liver lesions (33); and e) while 2-OADC is ubiquitous and may be seen throughout the body, inflammatory response is confined to BECs and salivary epithelial cells (SECs) (27,34).
The fact that the autoimmune response specifically targets BECs and SECs suggests that these cells must have some peculiarities. One of these may be that these cells, in contrast to others, aberrantly express PDC-E2 on its apical surface (35-37). This is an early phenomenon during PBC, one that develops much earlier than class-II HLA molecules (38). The reason why PDC-E2 is expressed on cell surfaces is not clearly understood. This occurs despite these cells lacking PDC-E2 messenger RNA (mRNA) (39). Hence these proteins must come from the extracellular environment, including the bile (40), or other phagocytized cells (41,42). Others have mentioned that PDC-E2 is released by mitochondria into the cytoplasm during apoptosis, but self-reactive epitopes are present on the surface of still intact cells (43). BECs have been seen to phagocytize apoptotic cells (44). Their presence as a consequence of apoptotic cell phagocytosis is of particular interest since in the course of this process cell molecules, including PDC-E2 components (45), experience changes leading to neoantigenic peptide formation (46-49). Despite the changes that PDC-E2 may undergo during apoptosis, epitopes that are recognized by AMA and self-reactive T and B cells remain in apoptotic BECs (44,50). This is because PDC-E2 experiences no lipoil domain glutathiolation in these cells, thus escapes degradation, and can then be expressed on the cell's surface. In cells other than BECs and SECs these proteins are catabolized in a different manner -PDC-E2 undergoes glutathiolation, experiences degradation by apoptosis-related enzymes, and disappears as an autoantigen.
Apical staining, as seen in BECs from patients with PBC, results from complex formation between IgA AMA and PDC-E2, which suggests that IgA may play a role in the destruction of small bile ducts. Regarding this, a PDC-E2-specific monoclonal IgA has been seen to enter BECs via the polymeric immunoglobulin receptor (pIgR); there it binds PDC-E2's catalytic component (51), forms complexes with it, and inactivates it (52-55). In this way AMA may induce mitochondrial dysfunction and apoptosis (56). The fact that BEC incubation with serum from patients with PBC results in caspase activation and cell apoptosis advocates for this notion (57). BEC-bound AMA probably have a dual origin. On the one hand they may originate in the bile; on the other, they may have been synthesized by B cells within portal spaces. In fact, the bile of patients with PBC contains IgG and IgA with anti-PDC-E2 activity (58), and 10% of B cells present in portal spaces release PDC-reactive antibodies (59).
Against the pathogenetic role of PDC-E2 in the BECs of patients with PBC stands the fact that transgenic mice expressing PDC-E2 on the surface of BECs have no liver or biliary lesions (60). This suggests that developing lesions require, in addition to an aberrant expression of PDC-E2 on the cell's surface, some molecular change in this complex, whether caused by xenobiotics, microorganisms (61,62), or apoptosis. During the latter some autoantigens, usually tolerated by the body, experience small molecular changes that render them intolerable to the immune system, which responds with aggression against cells harboring them.
Cell-mediated immunity
While the role of humoral immunity in the pathogenesis of PBC lesions remains unclear, evidence suggests that T cells play a key role in BEC and SEC death, and in bile and salivary duct destruction (3,10,20,63). Since early during the disease portal spaces of patients with PBC are infiltrated with B, T and plasma cells (20,64), and involved bile duct walls are infiltrated with intraepithelial CD8+ y CD4+, particularly CD4+CD28- (65,66), lymphocytes reactive to PDC-E2 (3,19,20,31,64,67) and native human antigen (68,69). While activated CD4+ cells (CD45RO) specifically recognize peptide PDC-E2163-176 (20,70,71), CD8+ cells in these patients recognize the PDC-E2159-167 epitope (3,63). As can be seen both cell types are reactive to the same amino-acid sequences or to nearby sequences in the lipoil domain.
CD4+CD28- T cells are believed to potentially play a role in the pathogenesis of autoimmune diseases such as PBC (65). These cells are highly increased in these conditions, express high amounts of IFNγ (72,73), are self-reactive and cytolytic, and survive many years because of their excessively expressing Bcl-2 and resistance to apoptosis (74).
Both the adaptive and innate autoimmune responses are regulated by two distinct CD4+ T cell types - Th1 and Th2. Th1 cells produce proinflammatory cytokines (IL2, IFNγ, TNFα), help cytotoxic CD8+ cells, regulate cell-mediated immune response, and activate NK cells. Th2 cells produce anti-inflammatory cytokines (IL4, IL5, IL6, IL10 e IL13), influence humoral response, and impact the differentiation and activation of B cells into plasma cells. In PBC immune response is Th1 in type (75-78). Indeed, monocucleated cells near small bile ducts express IFNγ mRNA, which correlates to portal inflammatory activity (40). In addition, peripheral blood mononuclear cells (PBMCs) in patients with PBC preferentially synthesize Th1 cytokines (79,80).
Inflammatory cell subpopulations infiltrating tissues mostly depend on chemokines released by altered cells, since these cytokines attract lymphocytes fitted with their receptors. The plasma and portal spaces of patients with PBC have revealed high protein 10 (CXCL10), monokine MIG (CXCL9), and fractalkine (CX3CL1) levels. While the former two are synthesized by macrophages exposed to IFNγ, the latter is produced by BECs in response to IFNγ and TNFα (81,82). While the CX3CL1 receptor may be found in CD4+ and CD8+ lymphocytes, CXCR3 and CCR5 receptors are preferentially expressed by Th1 cells (83,84), and CCR3 and CCR4 receptors by Th2 cells (83). Early during PBC mononuclear cells in portal infiltrates and around bile ducts have CXCR3 and CX3CL1 receptors (85), which accounts for the Th1 CD4+ cells present in this disease. Also osteopontin, a molecule that contributes to mononuclear cell recruitment and granuloma formation, abounds in portal spaces of patients with PBC (86).
Portal spaces and spaces between epithelial cells also contain CD8+ lymphocytes that play a role in the degeneration and death of BECs with aberrant expression of PDC-E2 and class-I and -II HLA molecules (3,87-91). Other cells also found in the biliary epithelium of interlobular ducts include CD20+ (B) cells, which advocates for a role of humoral immunity in the pathogenesis of PBC (92). This should not be surprising when consideration is given to the inter-relation between B and T cells, regarding the fact that the latter favor the former's proliferation (93).
CD4+CD25 regulatory T cells (Treg) play a key role in the prevention of T-cell-mediated autoimmune conditions (94,95), and their numbers and function decrease following the loss of immune tolerance and immune aggression. The molecule Foxp3 (Forhead P3) is a Treg function marker (95-98). Its detection in biopsies from patients with PBC shows, in contrast to other liver conditions, that they are very scarce in portal spaces (99). Experimental evidence suggests that this reduction of Treg cells plays a role in the pathogenesis of PBC. Function of these cells may be delayed in mice by blocking TGFβRII receptors, which are specific Treg g lymphocytes. Mice with this blockade develop portal infiltration with CD4+ and CD8+ cells, bile duct destruction, high AMA titers, and elevated IFNγ and TNFα levels (100-102). This laboratory model also demonstrates the importance of the TGFβ pathway for the prevention of autoimmune response. This growth factor conditions the suppressive capacity of Treg cells as it is required for Foxp3 synthesis in these cells (103). The cause of this Treg decrease is unknown but may be genetic in origin, as it is also found in first-degree female relatives. Patients with PBC have been seen to exhibit a low expression of the AE2 anionic exchanger gene (104,105) - AE2 plays a role in intracellular pH regulation (106) through bicarbonate secretion. When this exchanger's activity is low cells - including lymphocytes - become alkaline, and their activity decreases (107). AE2-deficient mice develop lesions similar to those of primary biliary cirrhosis - PDC-E2-specific AMA appear, the CD8+ population expands, and Treg numbers decline (108). A genetic defect involving AE2 may modify the immune response and result in the development of PBC-related lesions.
Innate immunity
Innate immunity represents a first-line defensive system that becomes immediately activated against micro-organisms. Its cellular component -macrophages, dendritic cells (DCs), antigen-presenting cells (APCs), NK cells- determines adaptive immune response quantity and quality, including T- and B-cell response and antibody production (109-111). These cells play a dual role - on the one hand they stimulate B- and T-cell response against infection, on the other hand they establish this cell tolerance to autoantigens, and hence prevent autoimmunity (112). In patients with PBC, this type of immunity is activated, reflected by the fact that exposure of these patients' PBMCs to CpG results in the production of AMA and various cytokines (IL-1β, IL6, IL8, TNFα) (113). TLRs (Toll-like receptors) play a role in innate immunity activation (114). These receptors are activated by some bacterial products, most particularly lipopolysaccharides (LPSs) and non-methylated oligonucleotides (CpG). TLR4 receptors, which are amply distributed in all tissues (115), start an innate immune response against gram-negative bacteria (116). In contrast to other liver conditions, in PBC, these receptors are vastly increased in the biliary epithelium and periportal hepatocytes, and, in them, expression extent correlates to disease stage (117). B-cell activation occurs via these receptors (118), hence increased IgM levels commonly found in patients with PBC are likely a memory B-cell response to their stimulus by bacterial substances. Exposure to these products may itself determine an increase in these receptors in PBC (119). Indeed, BEC stimulation with LPS increases TNFα production (120), which together with other proinflammatory cytokines (IFNγ) increases TLR4 expression on the cell surface (121,122). Other TLRs that may play a role in the pathogenesis of PBC include TLR9. These receptors have been thought to be activated in PBC as also occurs in various rheumatologic conditions (123,124). PBMC (125) or memory CD27+ B-cell (126,127) stimulation with CpG - a TLR9 ligand - in patients with PBC increases TLR8 and TLR9 expression in these cells, CD86 expression (monocytes, activated B cells), and IgM and AMA production.
Environmental factors
PBC risk factors include urinary tract infection, smoking, use of reproductive hormones, nail enamel (128), and living near a garbage dump (129). These environmental factors may initiate the autoimmune mechanisms that destroy small bile ducts (Fig. 1).
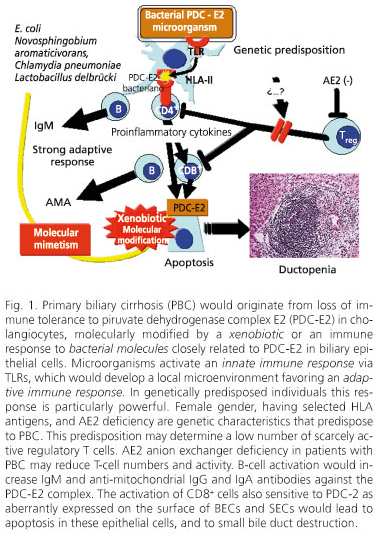
Numerous data suggest that some organisms or their products may play a role in the pathogenesis of PBC (130,131). Indeed, bacterial infection is common in these patients (132), and bacterial products have been consistently found in the liver tissue of patients with PBC (133,134). The mandatory passage of portal venous blood through the liver renders this organ a preferential target for bacterial products entering the body via this route. On passing through the liver micro-organisms or their products (135,136) would bind TLRs, activate innate immunity, and then activate adaptive immunity after the exposure of immune cells to bacterial peptides related to those in mitochondrial complexes (137).
Micro-organisms may play a role in the pathogenesis of PBC through several mechanims. We saw that their entry into the body starts innate immunity via TLRs. In addition, similarities between some bacterial and mitochondrial antigens (molecular mimetism) may determine loss of tolerance to one's own mitochondrial antigens (137-142). Peptide sequences in bacterial antigens that are similar to peptide sequences in mitochondrial products may be recognized by T-cell receptors and induce the latter's response to self antigens (143). It is possible that microbial infection disappears without a trace other than the presence of memory T cells capable of recognizing microbial antigens and mitochondrial self antigens.
Micro-organisms involved in the pathogenesis of PBC include Escherichia coli, some mycobacteria, Novosphingobium aromaticivorans, Chlamydia pneumonia, and Lactobacillus delbruecki, among others - Propioibacterium acnes (134), Lactobacillus delbrueckii (144), Azotobacter vinelandii, Pseudomonas putida, Helicobacter pylori, Streptococcus intermedius (145), and retroviruses -.
The involvement of Escherichia coli is based on many suspect observations. For instance, this bacterium has proteins similar to PDC-E2 (146); the serum and AMA of patients with PBC react with E. coli sequences that closely resemble epitope PDC-E2212-226 (141,146); feces from these patients contain R forms of E. coli that specifically react with AMA (146). Regarding the potential pathogenetic role of mycobacteria, AMA frequently develop during tuberculosis (147); lesions similar to those of PBC can be induced in mice through immunization with mycoplasma organisms (148), and cross reactivity has been found between protein HSP65 of Mycobacterium gordonae and epitope E2212-226 in PDC (149,150). Despite such observations the pathogenetic role of mycobacteria cannot be accepted yet because some of these observations could not be universally confirmed, and no such bacteria have been cultured in liver tissue thus far. The same goes for Chlamydia pneumoniae despite the fact that some investigators have reported the presence of this organism's messenger RNA (mRNA) and antigens in liver tissue (151). Others confirmed the presence of antibodies against C. pneumoniae in the serum of patients with PBC but failed to identify this organism's mRNA or antigens (152). While some consider cross reactivity to exist between the serum of patients with PBC and this bacterium, such reactivity is much lower than that seen with E. coli or N. aromaticivorans, and other authors never revealed it (153). Novosphingobium aromaticivorans has aroused special interest. This gram-negative bacterium is widely distributed in the soil and water. It has four lipoic acid domains highly homologous with human autoantigens. It has been seen that 100% of patients with PBC have antibody titers against N. aromaticivorans lipoic domains that are up to 1,000 times higher than those found against E. coli lipoic domains (154,155), and similar to those seen against PDC-E2 in these patients. This reactivity can be detected in asymptomatic PBC and in early-stage disease. Because of all this, this bacterium may possibly be directly involved in the pathogenesis of PBC. The retroviral etiology of PBC is based on the identification of viral particles and retroviral sequences within BECs in 75% of patients with PBC (156). In addition PDC-E2 is expressed by cholangiocytes following retroviral infection (157). Lastly, some have managed to improve this condition with lamivudine and zidovudine (158). Despite all this, the role of retroviral infection has not been demonstrated (159) as some authors have found no evidence for viral infection (160).
Xenobiotics, including xenobiotic drugs, may play a part in the pathogenesis of PBC given their capacity to modify mitochondrial proteins and induce an autoimmune response against them. B cells and T cells specific for these neoantigens may cross react with original mitochondrial proteins. The fact that AMA from patients with PBC react with new PDC-E2 epitopes generated by replacing lipoic acid in peptide PDC-E2E2173-184 with a number of similar synthetic structures does suggest so (161,162). Octinoic acid, usually found in perfumes, lipsticks, and food additives, is recognized by the serum from patients with PBC but not by the serum of control subjects or patients with other autoimmune diseases (161), and rabbits immunized with bromohexanoate ester-modified bovine serum albumin develop very high AMA titers that can inhibit PDC-E2; despite this, these animals do not develop PBC lesions (163,164). However, autoimmune cholangitis was induced when this was performed in Guinea pigs (165). All these findings support the idea that the exposure of predisposed subjects to selected xenobiotics may modify the immunogenic characteristics of PDC-E2 and induce PBC lesions.
Genetic factors
While the ultimate cause of PBC remains unknown, evidence suggests that genetic factors must play a role (166-168). This is pointed out by a 5% frequency of PBC among first-degree relatives of patients (169), which is 50 to 100 times higher than among the general population (170); relative risk among the offspring is 10.5 to 31.0, and it is 59 among daughters, and 10 among siblings (171). Concordance is 63% among homozygous twins (172). Furthermore, of all relatives of patients with PBC 2-13% are AMA carriers (171-175). Last, the disease is particularly common in selected geographical regions (176).
Given its association with other autoimmune conditions whether the HLA system can be related to PBC has been investigated. However, the role of class-I HLA antigens is uncertain (177,178). There is seemingly a closer relationship to class-II HLA molecules since an association of PBC with the DRB1*08 (DRB1*0801) allele has been commonly described among Caucasians (179,180), with DRB1*8 and DRB1*11 among Italians (181), and with DRB1*0803 among the Japanese (182,183). More recently an association was found for the DQA1*0401 allele and DR8-DQB1*0402 haplotype with disease progression (184). However, subsequent studies could not demonstrate this (178). On the other hand, DRB1*11 and DRB1*13 seem to have a protective role (180,185). Despite all this these associations, while relatively strong, only apply to a minority of patients, hence their seemingly secondary role. Regarding class-III HLA antigens results are also inconclusive. This region contains the gene coding for TNFα, but its relation to PBC is uncertain (186).
Other genetic factors that were studied to account for the high familial risk of having PBC include polymorphic varieties of immunomodulating molecules - chemokines, IL-1 (187), IL10 (188), vitamin D receptor (189), CTLA-4 (190,191), a T-cell activation inhibitor (192), vasoactive compound-producing enzymes, proteins involved in bile acid transportation and secretion (193), CYP2E1 (194), and IL-2α receptor (195) -. With the exception of vitamin D receptor polymorphisms, no clear association has been found between PBC and a specific gene to this day (166,178,196). Given the above, it is highly likely that PBC development requires the presence of genetic susceptibility, even if the latter does not by itself suffice to this end. This demands the presence of environmental factors to establish a loss of immune tolerance to autoantigens.
A genetic characteristic of PBC that cannot be ignored is that this disease affects almost exclusively women. The study of chromosome X has revealed that monosomy X is present in 5% of women with this disease, which is significantly higher than the frequency of this anomaly among age-matched women, namely 2% (197). A lost chromosome X may explain the preference of PBC for women (198). It has been suggested that this chromosome may contain a gene essential for immune regulation. In fact, autoimmune diseases, including PBC, are common in Turner's syndrome (XO) (199,200). Lastly, microchimerism, that is, fetal cells persisting in the maternal circulation after childbirth, may also account for the predominance of this disease in women. Its role is supported by the finding of fetal DNA in the liver of 42% of women with PBC many years after pregnancy (201). However, this hypothesis has not been corroborated by all (177), and is not believed to play a role in the pathogenesis of PBC (202,203).
References
1. Scheuer P. Primary biliary cirrhosis. Proc Roy Soc Med 1967; 60: 1257-60. [ Links ]
2. Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis). Wirchows Arch A 1978; 379: 103-12. [ Links ]
3. Kita H, Matsumura S, He X-S, Ansari AA, Lian ZX, Van de Water J, et al. Quantitative and functional analysis of PDC-E2 specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest 2002; 109: 1231-40. [ Links ]
4. Graham AM, Dollinger MM, Howie SE, Harrison DJ. Bile duct cells in primary biliary cirrhosis are 'primed' for apoptosis. Eur J Gastroenterol Hepatol 1998; 10: 553-7. [ Links ]
5. Tinmouth J, Lee M, Wanless IR, Tsui FW, Inman R, Heathcote EJ. Apoptosis of biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. Liver 2002; 22: 228-34. [ Links ]
6. Iwata M, Harada K, Kono N, Kaneko S, Kobayashi K, Nakanuma Y. Expression of Bcl-2 familial proteins is reduced in small bile duct lesions of primary biliary cirrhosis. Hum Pathol 2000; 31: 179-84. [ Links ]
7. Koga H, Sakisaka S, Ohishi M, Sata M, Tanikawa K. Nuclear DNA fragmentation and expression of Bcl-2 in primary biliary cirrhosis. Hepatology 1997; 25: 1077-84. [ Links ]
8. Dienes HP, Lohse AW, Gerken G, Schirmacher P, Gallati H, Lohr HF, Meyer zum Buschenfelde KH. Bile duct epithelia as target cells in primary biliary cirrhosis and primary sclerosing cholangitis. Virchows Arch 1997; 431: 119-24. [ Links ]
9. Harada K, Furubo S, Ozaki S, Hiramatsu K, Sudo Y, Nakanuma Y. Increased expression of WAF1 in intrahepatic bile ducts in primary biliary cirrhosis relates to apoptosis. J Hepatol 2001; 34: 500-6. [ Links ]
10. Harada K, Ozaki S, Gershwin ME, Nakanuma Y. Enhanced apoptosis relates to bile duct loss in primary biliary cirrhosis. Hepatology 1997; 26: 1399-405. [ Links ]
11. Harada K, Iwata M, Kono N, Koda W, Shimonishi T, Nakanuma Y. Distribution of apoptotic cells and expression of apoptosis related proteins along the intrahepatic biliary tree in normal and non-biliary diseased liver. Histopathology (Oxf) 2000; 37: 347-54. [ Links ]
12. Kuroki T, Seki S, Kawakita N, Nakatani K, Hisa T, Kitada T, Sakaguchi H. Expression of antigens related to apoptosis and cell proliferation in chronic nonsuppurative destructive cholangitis in primary biliary cirrhosis. Virchows Arch 1996; 429: 119-29. [ Links ]
13. Koga H, Sakisaka S, Ohishi M, Sata M, Tanikawa K Nuclear DNA fragmentation and expression of Bcl-2 in primary biliary cirrhosis. Hepatology 1997; 25: 1077-84. [ Links ]
14. Odin JA, Huebert RC, Casciola-Rosen L, LaRusso NF, Rosen A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest 2001; 108: 223-32. [ Links ]
15. Zheng SJ, Wang P, Tsabary G, Chen YH. Critical roles of TRAIL in hepatic cell death and hepatic inflammation. J Clin Invest 2004; 113: 58-64. [ Links ]
16. Spierings DC, de Vries EG, Vellenga E, van den Heuvel FA, Koornstra JJ, Wesseling J, et al. Tissue distribution of the death ligand TRAIL and its receptors. J Histochem Cytochem 2004; 52: 821-31. [ Links ]
17. Higuchi H, Grambihler A, Canbay A Bronk SF, Gores GJ. Bile acids up-regulate death receptor 5/TRAIL-receptor 2 expression via a c-Jun N-terminal kinase-dependent pathway involving Sp1. J Biol Chem 2004; 279: 51-60. [ Links ]
18. Iwata M, Harada K, Kono N, Kaneko S, Kobayashi K, Nakanuma Y. Expression of Bcl-2 familial proteins is reduced in small bile duct lesions of primary biliary cirrhosis. Hum Pathol 2000; 31: 179-84. [ Links ]
19. Kita H, Naidenko OV, Kronenberg M, Ansari AA, Rogers P, He XS, et al. Quantitation and phenotypic analysis of natural killer T cells in primary biliary cirrhosis using a human CD1d tetramer. Gastroenterology 2002; 123: 1031-43. [ Links ]
20. Shimoda S, Van de Water J, Ansari A, Nakamura M, Ishiashi H, Coppel Rl, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest 1998; 102: 1831-40. [ Links ]
21. Muntané J, Durán-Prado M, Rodríguez-Ariza A, Sánchez-Garrido MA, Collado JA, López-Sánchez LM, et al. Mechanisms of liver cell injury. Rev Esp Enferm Digest 2007; 99: 405-10. [ Links ]
22. Bogdanos DP, Baum H, Vergani D. Antimitochondrial and other autoantibodies. Clin Liver Dis 2003; 7: 759-77. [ Links ]
23. Mitchison H C, Bassendine M F, Hendrick A, Bennett MK, Bird G, Watson AJ, et al. Positive antimitochondrial antibody but normal alkaline phosphatase: is this primary biliary cirrhosis? Hepatology 1986; 6: 1279-84. [ Links ]
24. Metcalf J V, Mitchison H C, Palmer J M, Jones D E, Bassendine M F, James O F. Natural history of early primary biliary cirrhosis. Lancet 1996; 348: 1399-402. [ Links ]
25. Prince M, Chetwynd A, Newman W, Metcalf JV, James OF. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: follow-up for up to 28 years. Gastroenterology 2002; 123: 1044-51. [ Links ]
26. Oertelt S, Rieger R, Selmi C, Invernizzi P, Ansari AA, Coppel RL, et al. A sensitive bead assay for antimitochondrial antibodies: chipping away at AMA-negative primary biliary cirrhosis. Hepatology 2007; 45: 659-65. [ Links ]
27. Liu B, Shi XH, Zhang FC, Zhang W, Gao LX. Antimitochondrial antibody-negative primary biliary cirrhosis: a subsetof primary biliary cirrosis. Liver Internat 2008; 28: 233-9. [ Links ]
28. Bogdanos DP, Baum H, Vergani D. Antimitochondrial and other autoantibodies. Clin Liver Dis 2003; 7: 759-77. [ Links ]
toantibodies. Clin Liver Dis 2003; 7: 759-77.
29. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med 2005; 353: 1261-73. [ Links ]
30. Neuberger J. Primary biliary cirrhosis. Lancet 1997; 350: 875-9. [ Links ]
31. Van de Water J, Ansari AA, Surh CD, Coppel R, Roche T, Bonkovsky H, et al. Evidence for the targeting by 2-oxodehydrogenase enzymes in the T cell response of primary biliary cirrhosis. J. Immunol 1991; 146: 89-94. [ Links ]
32. Gershwin ME, Ansari AA, Mackay IR, Nakanuma Y, Nishio A, Rowley MJ, et al. Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev 2000; 174: 210-25. [ Links ]
33. Krams SM, Surh CD, Coppel RL, Ansari A, Ruebner B, Gershwin ME. Immunization of experimental animals with dihydrolipoamide acetyltransferase, as a purified recombinant polypeptide, generates mitochondrial antibodies but not primary biliary cirrhosis. Hepatology 1989; 9: 411-6. [ Links ]
34. Gershwin ME, Mackay IR. Primary biliary cirrhosis: paradigm or paradox for autoimmunity. Gastroenterology 1991; 100: 822-33. [ Links ]
35. Migliaccio C, Nishio A, Van de Water J, Ansari AA, Leung PS, Nakanuma Y, et al. Monoclonal antibodies to mitochondrial E2 components defines autoepitopes in primary biliary cirrhosis. J Immunol 1998; 127: 485-92. [ Links ]
36. Migliaccio C, Nishio A, Van De Water J, Ansari AA, Leung PS, Nakanuma Y, et al. Monoclonal antibodies to mitochondrial E2 components define autoepitopes in primary biliary cirrhosis, J Immunol 1998; 161: 5157-63. [ Links ]
37. Migliaccio C, Van De Water J, Ansari AA, Kaplan MM, Coppel RL, Lam KS, et al. Heterogeneous response of antimitochondrial autoantibodies and bile duct apical staining monoclonal antibodies to pyruvate dehydrogenase complex E2: the molecule versus the mimic, Hepatology 2001; 33: 792-801. [ Links ]
38. Tsuneyama K, Van de Water J, Leung PS, Cha S, Nakanuma Y, Kaplan M, et al. Abnormal expression of the E2 component of the pyruvate dehydrogenase complex on the luminal surface of biliary epithelium occurs before major histocompatibility complex class II and BB1/B7 expression. Hepatology 1995; 21: 1031-7. [ Links ]
39. Harada K, van de Water J, Leung PSC, Coppel R, Nakanuma Y, Gershwing ME. In situ nucleic acid hybridization of pyruvate deshydrogenasecomplex-E2 in primary biliary cirrhosis: pyruvate deshydrogenase-E2 messenger RNA is expressed in hepatocytes but not in biliary epithelial cells. Hepatology 1997; 25: 27-32. [ Links ]
40. Harada K, van de Water J, Leung PS, Coppel RL, Ansari A, Nakanuma Y, et al. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominante of the Th1 subset. Hepatology 1997; 25: 791-6. [ Links ]
41. Monks J, Rosner D, Geske FJ, Lehman L, Hanson L, Neville MC, et al. Epithelial cells as phagocytes: apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release. Cell Death Differ 2005; 12: 107-14. [ Links ]
42. Travaglione S, Falzano L, Fabbri A, Stringaro A, Fais S, Fiorentini C. Epithelial cells and expression of the phagocytic marker CD68: scavenging of apoptotic bodies following Rho activation. Toxicol In Vitro 2002; 16: 405-11. [ Links ]
43. Macdonald P, Palmer J, Kirby JA, Jones DE. Apoptosis as a mechanism for cell surface expression of the autoantigen pyruvate dehydrogenase complex. Clin Exp Immunol 2004; 136: 559-67. [ Links ]
44. Allina J, Hu B, Sullivan DM, Fiel MI, Thung SN, Bronk SF, et al. T cell targeting and phagocytosis of apoptotic biliary epithelial cells in primary biliary cirrhosis. J Autoimmun 2006; 27: 232-41. [ Links ]
45. Matsumura S, Van De Water J, Kita H, Coppel RL, Tsuji T, Yamamoto K, et al. Contribution to antimitochondrial antibody production: cleavage of pyruvate dehydrogenase complex-E2 by apoptosis related proteases. Hepatology 2002; 35: 14-22. [ Links ]
46. Rosen A, Casciola-Rosen L. Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ 1999; 6: 6-12 . [ Links ]
47. Utz PJ, Hottelet M, Schur PH, Anderson P. Proteins phosphorylated during stress-induced apoptosis are common targets for autoantibody production in patients with systemic lupus erythematosus. J Exp Med 1997; 185: 843-54. [ Links ]
48. Casciola-Rosen L, Rosen A, Petri M, Schlissel M. Surface blebs on apoptotic cells are sites of enhanced procoagulant activity: implications for coagulation events and antigenic spread in systemic lupus erythematosus. Proc Natl Acad Sci U S A 1996; 93: 1624-9. [ Links ]
49. Bellone M. Apoptosis, cross-presentation, and the fate of the antigen specific immune response. Apoptosis 2000; 5: 307-14. [ Links ]
50. Macdonald P, Palmer J, Kirby JA, Jones DE. Apoptosis as a mechanism for cell surface expression of the autoantigen pyruvate dehydrogenase complex. Clin Exp Immunol 2004; 136: 559-67. [ Links ]
51. Fussey SP, Bassendine MF, Fittes D, Turner IB, James OF, Yeaman SJ. The E1 alpha and beta subunits of the pyruvate dehydrogenase complex are M2'd' and M2'e' autoantigens in primary biliary cirrhosis. Clin Sci (Lond) 1989; 77: 365-8. [ Links ]
52. Fukushima N, Nalbandian G, Van De Water J, White K, Ansari AA, Leung P, et al. Characterization of recombinant monoclonal IgA anti-PDC-E2 autoantibodies derived from patients with PBC, Hepatology 2002; 36: 1383-92. [ Links ]
53. Malmborg AC, Shultz DB, Luton F, Mostov KE, Richly E, Leung PS, et al. Penetration and co-localization in MDCK cell mitochondria of IgA derived from patients with primary biliary cirrhosis, J Autoimmun 1998; 11: 573-80. [ Links ]
54. Teoh KL, Rowley MJ, Zafirakis H, Dickson ER, Wiesner RH, Gershwin ME, MacKay IR. Enzyme inhibitory autoantibodies to pyruvate dehydrogenase complex in primary biliary cirrhosis: applications of a semiautomated assay. Hepatology 1994; 20: 1220-4. [ Links ]
55. Teoh KL, Mackay IR, Rowley MJ, Fussey SP. Enzyme inhibitory autoantibodies to pyruvate dehydrogenase complex in primary biliary cirrhosis differ for mammalian, yeast and bacterial enzymes: implications for molecular mimicry. Hepatology 1994; 19: 1029-33. [ Links ]
56. Masuda J, Omagari K, Ohba K, Hazama H, Kadokawa Y, Kinoshita H, et al. Correlation between histopathological findings of the liver and IgA class antibodies to 2-oxo-acid dehydrogenase complex in primary biliary cirrhosis. Dig Dis Sci 2003; 48: 932-8. [ Links ]
57. Matsumura S, Van De Water J, Leung P, Odin JA, Yamamoto K, Gores GJ, et al. Caspase induction by IgA antimitochondrial antibody: IgA-mediated biliary injury in primary biliary cirrhosis, Hepatology 2004; 39: 1415-22. [ Links ]
58. Nishio A, van de Water J, Leung PS, Joplin R, Neuberger JM, Lake J, et al. Comparative studies of antimitochondrial autoantibodies in sera and bilis in primary biliary cirrhosis. Hepatology 1997; 25: 1085-9. [ Links ]
59. Bjorkland A, Loof L, Mendel-Hartvig I, Totterman TH. Primary biliary cirrhosis. High proportions of B cells in blood and liver tissue produce anti-mitochondrial antibodies of several Ig classes. J Immunol 1994; 153: 2750-7. [ Links ]
60. Inamura K, Tsuji H, Nakamoto Y, Suzuki M, Kaneko S. Transgenic mice aberrantly expressing pyruvate dehydrogenase complex E2 component on biliary epithelial cells do not show primary biliary cirrhosis. Clin Exp Immunol 2006; 145: 93-100. [ Links ]
61. Bruggraber SF, Leung PS, Amano K, Quan C, Kurth MJ, Nantz MH, et al. Autoreactivity to lipoate and a conjugated form of lipoate in primary biliary cirrhosis. Gastroenterology 2003; 125: 1705-13 [ Links ]
62. Amano K, Leung PS, Xu Q, Marik J, Quan C, Kurth MJ, et al. Xenobiotic-induced loss of tolerance in rabbits to the mitochondrial autoantigen of primary biliary cirrhosis is reversible. J Immunol 2004; 172: 6444-52. [ Links ]
63. Kita H, Lian ZX, Van de Water J, He XS, Matsumura S, Kaplan M, et al. Identification of HLA-A2-restricted CD8(þ) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J Exp Med 2002; 195: 113-23. [ Links ]
64. Jones DE, Palmer JM, James OF, Yeaman SJ, Bassendine MF, Diamond AG. T-cell responses to the components of pyruvate dehydrogenase complex in primary biliary cirrhosis. Hepatology 1995; 21: 995-1002. [ Links ]
65. Isse K, Harada K, Sato Y, Nakanuma Y. Characterization of biliary intra-epithelial lymphocytes at different anatomical levels of intrahepatic bile ducts under normal and pathological conditions: numbers of CD4CD28- intra-epithelial lymphocytes are increased in primary biliary cirrhosis. Pathol Int 2006; 56: 17-24. [ Links ]
66. Kamihira T, Shimoda S, Harada K, Kawano A, Handa M, Baba E, et al. Distinct costimulation dependent and independent autoreactive T-cell clones in primary biliary cirrhosis. Gastroenterology 2003; 125: 1379-87. [ Links ]
67. Van de Water J, Ansari A, Prindiville T, Coppel RL, Ricalton N, Kotzin BL, et al. Heterogeneity of autoreactive T cell clones specific for the E2 component of the pyruvate dehydrogenase complex in primary biliary cirrhosis. J Exp Med 1995; 181: 723-33. [ Links ]
68. Jones DE, Palmer JM, Yeaman SJ, Bassendine MF, Diamond AG. T cell responses to natural human proteins in primary biliary cirrhosis. Clin Exp Immunol 1997; 107: 562-8. [ Links ]
69. Akbar SM, Yamamoto K, Miyakawa H, Ninomiya T, Abe M, Hiasa Y, et al. Peripheral blood T-cell responses to pyruvate dehydrogenase complex in primary biliary cirrhosis: role of antigen-presenting dendritic cells. Eur J Clin Invest 2001; 31: 639-46. [ Links ]
70. Shigematsu H, Shimoda S, Nakamura M, Matsushita S, Nishimura Y, Sakamoto et al. Fine specificity of T cells reactive to human PDC-E2 163-176 peptide, the immunodominant autoantigen in primary biliary cirrhosis: implications for molecular mimicry and cross-recognition among mitochondrial autoantigens. Hepatology 2000; 32: 901-9. [ Links ]
71. Shimoda S, Nakamura M, Shigematsu H, Tanimoto H, Gushima T, Gershwin ME, Ishibashi H. Mimicry peptides of human PDC-E2 163-176 peptide, the immunodominant T-cell epitope of primary biliary cirrhosis. Hepatology 2000; 31: 1212-6. [ Links ]
72. Namekawa T, Snyder MR, Yen JH Goehring BE, Leibson PJ, Weyand CM, et al. Killer cell activating receptors function as costimulatory molecules on CD4+CD28null T cells clonally expanded in rheumatoid arthritis. J Immunol 2000; 165: 1138-45. [ Links ]
73. Weyand CM, Klimiuk PA, Goronzy JJ. Heterogeneity of rheumatoid arthritis: From phenotypes to genotypes. Springer Semin Immunopathol 1998; 20: 5-22. [ Links ]
74. Schirmer M, Vallejo AN, Weyand CM, Goronzy JJ. Resistance to apoptosis and elevated expression of Bcl-2 in clonally expanded CD4+CD28- T cells from rheumatoid arthritis patients. J Immunol 1998; 161: 1018-25. [ Links ]
75. Berg PA, Klein R, Rocken M. Cytokines in primary biliary cirrhosis. Semin Liver Dis 1997; 17: 115-23. [ Links ]
76. Martínez OM, Villanueva JC, Gershwin ME, Krams SM Cytokine patterns and cytotoxic mediators in primary biliary cirrhosis. Hepatology 1995; 21: 113-9. [ Links ]
77. Harada K, Isse K, Kamihira T, Shimoda S, Nakanuma Y. Th1 cytokine-induced downregulation of PPARgamma in human biliary cells relates to cholangitis in primary biliary cirrhosis. Hepatology 2005; 41: 1329-38. [ Links ]
78. Lohr HF, Schlaak JF, Gerken G, Fleischer B, Dienes HP, Meyer zum Buschenfelde KH Phenotypical analysis and cytokine release of liver-infiltrating and peripheral blood T lymphocytes from patients with chronic hepatitis of different etiology. Liver 1994; 14: 161-6. [ Links ]
79. Sekiya H, Komatsu T, Isono E, Furukawa M, Matsushima S, Yamaguchi N, et al. Decrease in the prevalence of IL-4-producing CD4T cells in patients with advanced stage of primary biliary cirrhosis. Am J Gastroenterol 1999; 94: 3589-94. [ Links ]
80. Nagano T, Yamamoto K, Matsumoto S, Okamoto R, Tagashira M, Ibuki N, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol 1999; 19: 422-7. [ Links ]
81. Muehlhoefer A, Saubermann LJ, Gu X, Luedtke-Heckenkamp K, Xavier R, Blumberg RS, et al. Fractalkine is an epithelial and endothelial cell-derived chemoattractant for intraepithelial lymphocytes in the small intestinal mucosa. J Immunol 2000; 164: 3368-76. [ Links ]
82. Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med 1998; 188: 1413-9. [ Links ]
83. Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med 1998; 187: 875-83. [ Links ]
84. Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest 1998; 101: 746-54. [ Links ]
85. Harada K, Tsuneyama K, Yasoshima M, Kanemori Y, Ohta H, Masuda S, et al. Type 1 and type 2 memory T cells imbalance shown by expression of intrahepatic chemokine receptors relates to pathogenesis of primary biliary cirrhosis. Hepatol Res 2002; 24: 290-9. [ Links ]
86. Harada K, Ozaki S, Sudo Y, Tsuneyama K, Ohta H, Nakanuma Y. Osteopontin is involved in the formation of epithelioid granuloma and bile duct injury in primary biliary cirrhosis. Pathol Int 2003; 53: 8-17. [ Links ]
87. Kita H, Imawari M, Gershwin ME. Cellular immune response in primary biliary cirrhosis. Hepatol Res 2004; 28: 12-7. [ Links ]
88. Ohtsuka K, Iiai T, Watanabe H, Tanaka T, Miyasaka M, Sato K, et al. Similarities and differences between extrathymic T cells residing in mouse liver and intestine. Cell Immunol 1994; 153: 52-66. [ Links ]
89. Matsumura S, Kita H, He XS, Ansari AA, Lian ZX, Van De Water J, et al. Comprehensive mapping of HLA-A0201-restricted CD8 T-cell epitopes on PDC-E2 in primary biliary cirrhosis. Hepatology 2002; 36: 1125-34. [ Links ]
90. Nishimoto H, Yamada G, Mizuno M, Tsuji T. Immunoelectron microscopic localization of MHC class 1 and 2 antigens on bile duct epithelial cells in patients with primary biliary cirrhosis. Acta Med Okayama 1994; 48: 317-22. [ Links ]
91. Van den Oord JJ, Sciot R, Desmet VJ. Expression of MHC products by normal and abnormal bile duct epithelium. J Hepatol 1986; 3: 310-7. [ Links ]
92. Nakanuma Y. Distribution of B lymphocytes in nonsuppurative cholangitis in primary biliary cirrhosis. Hepatology 1993; 18: 570-5 [ Links ]
93. Beagley KW, Husband AJ. Intraepithelial lymphocytes: origins, distribution, and function. Crit Rev Immunol 1998; 18:237-54. [ Links ]
94. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol 2004; 22: 531-62. [ Links ]
95. Fehervari Z, Sakaguchi S. CD4+Tregs and immune control, J Clin Invest 2004; 114: 1209-17. [ Links ]
96. Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol 2005; 6: 345-52. [ Links ]
97. Sakaguchi S, Setoguchi R, Yagi H, Nomura T. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in self-tolerance and autoimmune disease. Curr Top Microbiol Immunol 2006; 305: 51-66. [ Links ]
98. Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest 2006; 116: 1713-22. [ Links ]
99. Lan RY, Cheng C, Lian ZX, Tsuneyama K, Yang GX, Moritoki Y, et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis, Hepatology 2006; 43: 729-37. [ Links ]
100. Oertelt S, Lian Z-X, Cheng C-M, Chuang Y-H, Padgett KA, He X-S, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol 2006; 177: 1655-60. [ Links ]
101. Shindo M, Mullin GE, Braun-Elwert L, Bergasa NV, Jones EA, James SP. Cytokine mRNA expression in the liver of patients with primary biliary cirrhosis (PBC) and chronic hepatitis B (CHB). Clin Exp Immunol 1996; 105: 254-9. [ Links ]
102. Yasoshima M, Kono N, Sugawara H, Katayanagi K, Harada K, Nakanuma Y. Increased expression of interleukin-6 and tumor necrosis factor-alpha in pathologic biliary epithelial cells: in situ and culture study. Lab Invest 1998; 78: 89-100. [ Links ]
103. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med 2005; 201:1061-7. [ Links ]
104. Prieto J, Qian C, García N, Díez J, Medina JF. Abnormal expression of anion exchanger genes in primary biliary cirrhosis. Gastroenterology 1993; 105: 572-8. [ Links ]
105. Melero S, Spirli C, Zsembery A, Medina JF, Joplin RE, Duner E, et al. Defective regulation of cholangiocyte Cl-/HCO3(-) and Na+/H+ exchanger activities in primary biliary cirrhosis. Hepatology 2002; 35: 1513-21. [ Links ]
106. Alper SL. Molecular physiology of SLC4 anion exchangers. Exp Physiol 2006; 91: 153-61. [ Links ]
107. Lardner A. The effects of extracellular pH on immune function. J Leukoc Biol 2001; 69: 522-30. [ Links ]
108. Salas JT, Banales JM, Sarvide S, Recalde S, Ferrer A, Uriarte I, et al. Ae2a,B-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology 2008; 134: 1482-93. [ Links ]
109. Medzhitov R, Janeway CA Jr. Innate immunity: impact on the adaptive immune response. Curr Opin Immunol 1997; 9: 4-9. [ Links ]
110. Zitvogel L. Dendritic and natural killer cells cooperate in the control/switch of innate immunity. J Exp Med 2002; 195: F9-F14. [ Links ]
111. Moretta A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat Rev Immunol 2002; 2: 957-64. [ Links ]
112. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol 2000; 18: 767-811. [ Links ]
113. Mao TK, Lian ZX, Selmi C, Ichiki Y, Ashwood P, Ansari AA, et al. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology 2005; 42: 802-8. [ Links ]
114. Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature 2000; 406: 782-7. [ Links ]
115. Pasterkamp G, Van Keulen JK, De Kleijn DP. Role of toll-like receptor 4 in the initiation and progression of atherosclerotic disease. Eur J Clin Invest 2004; 34: 328-34. [ Links ]
116. Beutler B. TLR4: central component of the sole mammalian LPS sensor. Curr Opin Immunol 2000; 12: 20-6. [ Links ]
117. Wang A-P, MigitaK, Ito M, Takii Y, Daikoku M, Yokoyama T, et al. Hepatic expression of toll-like receptor 4 in primar biliary cirrhosis Journal of Autoimmunity 2005; 25: 85-91. [ Links ]
118. Pasare C, Medzhitov R. Control of B-cell responses by toll-like receptors. Nature 2005; 438: 364-8. [ Links ]
119. Romics Jr L, Dolganiuc A, Kodys K, Drechsler Y, Oak S, Velayudham A, et al. Selective priming to toll-like receptor 4 (TLR4), not TLR2, ligands by P. acnes involves up-regulation of MD-2 in mice. Hepatology 2004; 40: 555-64. [ Links ]
120. Harada K, Ohira S, Isse K, Ozaki S, Zen Y, Sato Y, et al. Lipopolysaccharide activates nuclear factor-kappaB through toll-like receptors and related molecules in cultured biliary epithelial cells. Lab Invest 2003; 83: 1657-67. [ Links ]
121. Wolfs TG, Buurman WA, van Schadewijk A, de Vries B, Daemen MA, Hiemstra PS, et al. In vivo expression of toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNFalpha mediated up-regulation during inflammation. J Immunol 2002; 168: 1286-93. [ Links ]
122. Faure E, Thomas L, Xu H, Medvedev A, Equils O, Arditi M. Bacterial lipopolysaccharide and IFN-gamma induce toll-like receptor 2 and toll-like receptor 4 expression in human endothelial cells: role of NF-kappa B activation. J Immunol 2001; 166: 2018-24. [ Links ]
123. Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 2006; 203: 553-61. [ Links ]
124. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature 2002; 416: 603-7. [ Links ]
125. Moritoki Y, Lian ZX, Wulff H, Yang GX, Chuang YH, Lan RY, et al. AMA Production in primary biliary cirrhosis is promoted by the TLR9 Ligand CpG and suppressed by potassium channel blockers. Hepatology 2007; 45: 314-22. [ Links ]
126. Kikuchi K, Lian ZX, Kimura Y, Selmi C, Yang GX, Gordon SC, et al. Genetic polymorphisms of toll-like receptor 9 influence the immune response to CpG and contribute to hyper-IgM in primary biliary cirrhosis. J Autoimmun 2005; 24: 347-52. [ Links ]
127. Kikuchi K, Lian ZX, Yang GX, Ansari AA, Ikehara S, Kaplan M, et al. Bacterial CpG induces hyper-IgM production in CD27(+) memory B cells in primary biliary cirrhosis. Gastroenterology 2005; 128: 304-12. [ Links ]
128. Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology 2005; 42: 1194-202. [ Links ]
129. Ala A, Stanca CM, Bu-Ghanim M, Ahmado I, Branch AD, Schiano TD, Odin JA, Bach N: Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology 2006; 43: 525-31. [ Links ]
130. Galperin C, Gershwin ME. Immunopathogenesis of gastrointestinal and hepatobiliary diseases. JAMA 1997; 278: 1946-55. [ Links ]
131. Sakisaka S, Koga H, Sasatomi K, Mimura Y, Kawaguchi T, Tanikawa K. Biliary secretion of endotoxin and pathogenesis of primary biliary cirrhosis. Yale J Biol Med 1997; 70: 403-8. [ Links ]
132. Butler P, Valle F, Hamilton-Miller JM, Brumfitt W, Baum H, Burroughs AK. M2 mitochondrial antibodies and urinary rough mutant bacteria in patients with primary biliary cirrhosis and in patients with recurrent bacteriuria. J Hepatol 1993; 17: 408-14. [ Links ]
133. Haydon GH, Neuberger J. PBC: an infectious disease? Gut 2000; 47: 586-8. [ Links ]
134. Harada K, Tsuneyama K, Sudo Y, Masuda S, Nakanuma Y. Molecular identification of bacterial 16S ribosomal RNA gene in liver tissue of primary biliary cirrhosis: is Propionibacterium acnes involved in granuloma formation? Hepatology 2001; 33: 530-6. [ Links ]
135. Freudenberg MA, Galanos C. Bacterial lipopolysaccharides: structure, metabolism and mechanisms of action. Int Rev Immunol 1990; 6: 207-21. [ Links ]
136. Nolan JP. Endotoxin, reticuloendothelial function, and liver injury. Hepatology 1981; 1: 458-65. [ Links ]
137. Selmi C, Gershwin EM. Bacteria and human autoimmunity: the case of primary biliary cirrhosis. Curr Opin Rheumatol 2004; 16: 406-10. [ Links ]
138. Leung PS, Coppel RL, Gershwin ME. Etiology of primarybiliary cirrhosis: the search for the culprit. Semin Liver Dis 2005; 25: 327-36. [ Links ]
139. Long SA, Van de Water J, Gershwin ME. Antimitochondrial antibodies in primary biliary cirrhosis: the role of xenobiotics. Autoimmun Rev 2002; 1: 37-42. [ Links ]
140. Kita H, He XS, Gershwin ME. Autoimmunity and environmental factors in the pathogenesis of primary biliary cirrhosis. Ann Med 2004; 36: 72-80. [ Links ]
141. Bogdanos DP, Baum H, Grasso A, Okamoto M, Butler P, Ma Y, et al. Microbial mimics are major targets of crossreactivity with human pyruvate dehydrogenase in primary biliary cirrhosis. J Hepatol 2004; 40: 31-9. [ Links ]
142. Bogdanos DP, Baum H, Gunsar F, Arioli D, Polymeros D, Ma Y, et al. Extensive homology between the major immunodominant mitochondrial antigen in primary biliary cirrhosis and Helicobacter pylori does not lead to immunological cross-reactivity. Scand J Gastroenterol 2004; 39: 981-7. [ Links ]
143. Evavold BD, Allen PM. Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand, Science 1991; 252: 1308-10. [ Links ]
144. Bogdanos DP, Baum H, Okamoto M, Montalto P, Sharma UC, Rigopoulou EI, et al. Primary biliary cirrhosis is characterized by IgG3 antibodies cross-reactive with the major mitochondrial autoepitope and its Lactobacillus mimic. Hepatology 2005; 42: 458-65. [ Links ]
145. Haruta I, Kikuchi K, Hashimoto E, Kato H, Hirota K, Kobayashi M, et al. A possible role of histone-like DNA-binding protein of Streptococcus intermedius in the pathogenesis of bileduct damage in primary biliary cirrosis. Clinical Immunology 2008; 127: 245-51. [ Links ]
146. Hopf U, Moller B, Stemerowicz R, Lobeck H, Rodloff A, Freudenberg M, et al. Relation between Escherichia coli R (rough)-forms in gut, lipid A in liver, and primary biliary cirrhosis. Lancet 1989; ii: 1419-22. [ Links ]
147. Klein R, Berg PA. Demostration of antibodies against the pyruvate dehydrogenase complex (M2) in sera from patients with tuberculosis. Hepatology 1992; 16: 556. [ Links ]
148. Johnson L, Wirostko E, Wirotsko W. Primary biliary cirrhosis in the mouse: induction by human micoplasma-like organism. Int J Exp Pathol 1990; 71: 701-12. [ Links ]
149. Bogdanos DP, Pares A, Baum H, Caballeria L, Rigopoulou EI, Ma Y, et al. Disease-specific cross-reactivity between mimicking peptides of heat shock protein of Mycobacterium gordonae and dominant epitope of E2 subunit of pyruvate dehydrogenase is common in Spanish but not British patients with primary biliary cirrhosis. J Autoimmun 2004; 22: 353-62. [ Links ]
150. Vilagut L, Vila J, Vinas O, Pares A, Gines A, Jimenez de Anta MT, Rodes J. Cross-reactivity of anti-Mycobacterium gordonae antibodies with the major mitochondrial autoantigens in primary biliary cirrhosis. J Hepatol 1994; 21: 673-7. [ Links ]
151. Abdulkarim AS, Petrovic LM, Kim WR, Angulo P, Lloyd RV, Lindor KD. Primary biliary cirrhosis: an infectious disease caused by Chlamydia pneumonie? J Hepatol 2004; 40: 380-4. [ Links ]
152. Leung PS, Park O, Matsumura S, Ansari AA, Coppel RL, Gershwin ME. Is there a relation between Chlamydia infection and primary biliary cirrhosis? Clin Dev Immunol 2003; 10: 227-33. [ Links ]
153. Liu H-Y, Deng A-M, Zhang J, Zhou Y, Yao D-K, Tu X-Q, et al. Correlation of Chlamydia pneumoniae infection with primary biliary cirrhosis. World J Gastroenterol 2005; 11: 4108-10. [ Links ]
154. Selmi C, Balkwill DL, Invernizzi P, Ansari AA, Coppel RL, Podda M, et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology 2003; 38: 1250-7. [ Links ]
155. Kaplan MM. Novosphingobium aromaticivorans: a potential initiator of primary biliary cirrhosis. Am J Gastroenterol 2004; 99: 2147-9. [ Links ]
156. Xu L, Sakalian M, Shen Z, Loss G, Neuberger J, Mason A. Cloning the human betaretrovirus proviral genome from patients with primary biliary cirrosis. Hepatology 2004; 39: 151-6. [ Links ]
157. Sutton I, Neuberger J. Primary biliary cirrhosis: seeking the silent partner of autoimmunity. Gut 2002; 50: 743-6. [ Links ]
158. Mason AL, Farr GH, Xu L, Hubscher SG, Neuberger JM. Pilot studies of single and combination antiretroviral therapy in patientes with primary biliary cirrhosis. Am J Gastroenterol 2004; 99: 2348-55. [ Links ]
159. Perron H, Seigneurin JM. Human retroviral sequences associated with extracellular particles in autoimmune diseases: epiphenomenon or possible role in aetiopathogenesis? Microbes Infect 1999; 1: 309-22. [ Links ]
160. Selmi C, Ross SR, Ansari AA, invernizzi P, Podda M, Coppel RL, Gershwing ME. Lack of immunobiological or molecular evidence for a role of mouse mammary tumor retrovirus in primary biliary cirrhosis. Gastroenterology 2004; 127: 493-501. [ Links ]
161. Amano K, Leung PS, Rieger R, Quan C, Wang X, Marik J, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol 2005; 174: 5874-83. [ Links ]
162. Leung SA, Quan C, Van De Water J, Nantz MH, Kurth MJ, Barsky D, et al. Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: connecting xenobiotics with primary biliary cirrhosis. J Immunol 2001; 167: 2956-63. [ Links ]
163. Leung PS, Quan C, Park C, Van De Water J, Kurth MJ, Nantz MH, et al. Immunization with a xenobiotic 6-bromohexanoate bovine serum albumin conjugate induces antimitochondrial antibodies. J Immunol 2003; 170: 5326-32. [ Links ]
164. Bruggraber SF, Leung PS, Amano K, Quan C, Kurth MJ, Nantz MH, et al. Autoreactivity to lipoate and a conjugated form of lipoate in primary biliary cirrhosis. Gastroenterology 2003; 125: 1705-13. [ Links ]
165. Leung PSC, Park O, Tsuneyama K, Kurth MJ, Lam KS, Ansari AA, et al. Induction of primary biliary cirrhosis in guinea pigs following chemical xenobiotic immunization. J Immunol 2007; 179: 2651-7. [ Links ]
166. Jones DEJ, Donaldson PT. Genetic factors in the pathogenesis of primary biliary cirrhosis. Clin Liver Dis 2003; 7: 841-64. [ Links ]
167. Donaldson PT. Genetics of liver disease: immunogenetics and disease pathogenesis. Gut 2004; 53: 599-608. [ Links ]
168. Tanaka A, Borchers AT, Ishibashi H, Ansari AA, Keen CL, Gershwin ME. Genetic and familial considerations of primary biliary cirrhosis. Am J Gastroenterol 2001; 96: 8-15. [ Links ]
169. Jones DE, Watt FE, Metcalf JV, Bassendine MF, James OF. Familial primary biliary cirrhosis reassessed: a geographically-based population study. J Hepatol 1999; 30: 402-7. [ Links ]
170. Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: convenient and inconvenient truths. Hepatology 2008; 47: 737-45. [ Links ]
171. Jones DEJ, Watt FE, Metcalf JV, Bassendine MF, James OFW. Familial primary biliary cirrhosis reassessed: a geographically-based population study. J Hepatol 1999; 30: 402-7. [ Links ]
172. Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernezzi P, Gish RG, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics and environment. Gastroenterology 2004; 127: 485-92. [ Links ]
173. Bach N, Schafner F. Familial primary biliary cirrhosis. J Hepatol 1994; 20: 698-701. [ Links ]
174. Brind AM, Bray GP, Portmann BC, Williams R. Prevalence and pattern of familial disease in primary biliary cirrhosis. Gut 1995; 36: 615-7. [ Links ]
175. Lazaridis KN, Juran BD, Boe GM, Slusser JP, Andrade M, Homburger HA, et al. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrosis. Hepatology 2007; 46: 785-92. [ Links ]
176. Prince MI, Chetwynd A, Diggle P, Jarner M, Metcalf JV, James OF. The geographical distribution of primary biliary cirrhosis is a well-defined cohort. Hepatology 2001; 34: 1083-8. [ Links ]
177. Invernizzi P, Miozzo M, Selmi C, Persani L, Battezzati PM, Zuin M, et al. X chromosome monosomy: a common mechanism for autoimmune diseases. J Immunol 2005; 175: 575-8. [ Links ]
178. Selmi C, Invernizzi P, Zuin M, Podda M, Seldin MF, Gershwin ME. Genes and (auto)immunity in primary biliary cirrosis. Genes Immun 2005; 6: 543-56. [ Links ]
179. Stone J, Wade JA, Cauch-Dudek K, Ng C, Lindor KD, Heathcote EJ. Human leucocyte antigen class II associations in serum antimitochondrial antibodies (AMA)-positive and AMA-negative primary biliary cirrhosis. J Hepatol 2002; 36: 8-13. [ Links ]
180. Donaldson PT, Baragiotta IB, Heneghan MA, Floreani A, Venturi C, Underhill JA et al. HLA Class II alleles, genotypes, haplotypes, and amino acids in primary biliary cirrhosis: a large-scale study. Hepatology 2006; 44: 667-74. [ Links ]
181. Invernizzi P, Selmi C, Poli F, Frison S, Floreani A, Alvaro D, et al. Human leukocyte antigen polymorphisms in Italian primary biliary cirrhosis: a multicenter study of 664 patients and 1992 healthy controls. Hepatology 2008; 48: 1906-12. [ Links ]
182. Maeda T, Onishi S, Saibara T, Iwasaki S, Yamamoto Y. HLA DRw8 and primary biliary cirrhosis. Gastroenterology 1992; 103: 118-9. [ Links ]
183. Seki T, Kiyosawa K, Ota M, Furuta S, Fukushima H, Tanaka E, et al. Association of primary biliary cirrhosis with human leukocyte antigen DPB1*0501 in Japanese patients. Hepatology 1993; 18: 73-8. [ Links ]
184. Donaldson P, Agarwal K, Craggs A, Craig W, Jones D, James O. HLA and interleukin-1 gene polymorphisms in primary biliary cirrhosis; associations with disease progression and disease susceptibility. Gut 2001; 48: 397-402. [ Links ]
185. Invernizzi P, Battezzati PM, Crosignani A, Peregro F, Poli F, Morabito A, et al. Peculiar HLA polymorphisms in Italian patients with primary biliary cirrhosis. J. Hepatol 2003; 38: 401-6. [ Links ]
186. Donaldson, PT. TNF gene polymorphisms in primary biliary cirrhosis: a critical appraisal. J Hepatol 1999; 31: 366-8. [ Links ]
187. Donaldson P, Agarwal K, Craggs A, Craig W, James O, Jones D. HLA and interleukin 1 gene polymorphisms in primary biliary cirrhosis: associations with disease progression and disease susceptibility. Gut 2001; 48: 397-402. [ Links ]
188. Matsushita M, Tanaka A, Kikuchi K Kitazawa E, Kawaguchi N, Kawashima Y, et al. Association of single nucleotide polymorphisms of the interleukin-10 promoter gene and susceptibility to primary biliary cirrhosis: immunogenetic differences in Italian and Japanese patients. Autoimmunity 2002; 35: 531-6. [ Links ]
189. Vogel A, Strassburg CP, Manns MP. Genetic association of vitamin D receptor polymorphisms with primary biliary cirrhosis and autoimmune hepatitis. Hepatology 2002; 35: 126-31. [ Links ]
190. Donaldson P, Veeramani S, Baragiotta A, Floreani A, Venturi C, Pearce S, et al. Cytototoxic T-lymphocyte-associated antigen-4 single nucleotide polymorphisms and haplotype in primary biliary cirrosis. Clin Gastroenterol Hepatol 2007; 6: 755-60. [ Links ]
191. Juran BD, Atkinson EJ, Schlicht EM, Fridley BL, Lazaridis KN. Primary biliary cirrhosis is associated with a genetic variant in the 3' flanking region of the CTLA4 gene. Gastroenterology 2008; 135: 1200-6. [ Links ]
192. Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev 2008; 223: 143-55. [ Links ]
193. Pauli-Magnus C, Kerb R, Fattinger K, Lang T, Anwald B, Kullak-Ublick GA, et al. BSEP and MDR3 haplotype structure in healthy Caucasians, primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology 2004; 39: 779-91. [ Links ]
194. Kimura Y, Selmi C, Leung PS, Mao TK, Schauer J, Watnik M, et al. Genetic polymorphisms influencing xenobiotic metabolism and transport in patients with primary biliary cirrhosis. Hepatology 2005; 41: 55-63. [ Links ]
195. Aoki CA, Roifman CM, Lian ZX, Bowlus CL, Norman GL, Shoenfeld Y, et al. IL-2 receptor alpha deficiency and features of primary biliary cirrhosis. J Autoimmun 2006; 27: 50-3. [ Links ]
196. Springer JE, Cole DE, Rubin LA, Cauch-Dudek K, Harewood L, Evrovski J, et al. Vitamin D-receptor genotypes as independent genetic predictors of decreased bone mineral density in primary biliary cirrhosis. Gastroenterology 2000; 118: 145-51. [ Links ]
197. Invernizzi P, Miozzo M, Battezzatti PM, Bianchi I, Grati FR, Simoni G, et al. The frequency of monosomy X in women with primary biliary cirrhosis. Lancet 2004; 363: 533-5. [ Links ]
198. Miozzo M, Selmi C, Gentilin B, Grati FR, Sirchia S, Oertelt S, et al. Prefential C chromosome loss but random inactivation characterize primary biliary cirrhosis. Hepatology 2007; 46: 456-62. [ Links ]
199. Elsheikh M, Wass J, Conway G. Autoimmune thyroid syndrome in women with Turner's syndrome-the association with karyotype. Clin Endocrinol (Oxf) 2001; 55: 223-6. [ Links ]
200. Milkiewicz P, Heathcote J: Primary biliary cirrhosis in a patient with Turner syndrome. Can J Gastroenterol 2005; 19: 631-3. [ Links ]
201. Fanning PA, Jonsson JR, Clouston AD, Edwards-Smith C, Balderson GA, Macdonald GA, et al. Detection of male DNA in the liver of famele patients with primary biliary cirrhosis. J Hepatol 2000; 33: 690-5. [ Links ]
202. Tanaka A, Lindor K, Gish R, Batts K, Shiratori Y, Omata M, et al. Fetal microchimerism alone does not contribute to the induction of primary biliary cirrhosis. Hepatology 1999; 30: 833-8. [ Links ]
203. Schoniger-Hekele M, Muller C, Ackermann J, Drach J, Wrba F, Penner E, et al. Lack of evidence for involvement of fetal microchimerism in patogenesis of primary biliary cirrhosis. Dig Dis Sci 2002; 47: 1909-14. [ Links ]
![]() Correspondence:
Correspondence:
José A. Solís herruzo.
Servicio de Medicina del Aparato Digestivo.
Hospital Universitario 12 de Octubre.
Ctra. de Andalucía, km. 5,400.
28041 Madrid, Spain.
e-mail: jsolis.hdco@salud.madrid.org
Received: 04-03-09.
Accepted: 08-03-09.

