










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.102 no.8 Madrid ago. 2010
Multiple non-metastatic gastrointestinal stromal tumors. Differential features
Tumores del estroma gastrointestinal múltiples no metastásicos. Aspectos diferenciales
M. Díaz Delgado1, A. Hernández Amate2, M. Sánchez León3, S. Pereira Gallardo3, E. Prieto Sánchez4, M. Jiménez Sáenz5 and R. González-Cámpora3
Department of Pathology. 1Hospital de Mérida. Mérida. Badajoz, Spain. 2Hospital Universitario Infanta Cristina. Badajoz, Spain. 3Hospital Universitario Virgen Macarena. Sevilla, Spain. 4Hospital de Motril. Granada, Spain. 5Servicio de Aparato Digestivo. Hospital Universitario Virgen Macarena. Sevilla, Spain
ABSTRACT
Introduction: gastrointestinal stromal tumors (GISTs) are specific, generally KIT (CD117)-positive, mesenchymal tumors of the digestive tract displaying KIT or PDGFRA gene mutations. Clinically, they tend to present as solitary tumors of the intestinal wall; more rarely, multiple tumors may occur in one or more organs.
Objective: to review the morphological, immunohistochemical and molecular features of multiple, non-metastatic forms of GIST.
Sources: review of the literature on Medline, and authors' own experience.
Conclusions: multiples GISTs may occur in three different contexts: as spontaneous lesions (in both adults and children); due to familial GIST syndrome (autosomal dominant inheritance); or in association with specific syndromes (e.g. Carney's triad, Carney-Stratakis syndrome, type I neurofibromatosis). Outside these contexts, the existence of multiple GISTs is deemed to be the result of tumor metastasis, and therefore indicative of advanced-stage disease. Clinicians need to be aware of these variants, whose prognosis and treatment differ.
Key words: Gastrointestinal stromal tumors. GIST. Carney's triad. Carney-Stratakis syndrome. Type I neurofibromatosis. Familial GIST syndrome. Pediatric GIST.
RESUMEN
Introducción: los tumores del estroma gastrointestinal (GIST) son neoplasias mesenquimales del tubo digestivo que generalmente expresan el receptor KIT (CD117) y muestran mutaciones en los genes KIT o PDGFRA. Aunque la forma de presentación clínica habitual es como una neoplasia mural solitaria, excepcionalmente pueden presentarse formas múltiples en el mismo o diferente órgano.
Objetivo: revisar las características morfológicas, inmunohistoquímicas y moleculares de las formas de GIST múltiples no metastásicos.
Fuentes: revisión de la literatura en Medline y la propia experiencia.
Conclusiones: los GIST múltiples pueden presentarse en tres contextos diferentes: lesiones espontáneas (del adulto o de la edad infantil); síndrome familiar propio (transmitido con herencia autosómica dominante); y lesiones asociadas a síndromes específicos (tríada de Carney, síndrome de Carney-Stratakis, y neurofibromatosis tipo I). Fuera de estos ámbitos, se interpreta que todo GIST múltiple es el resultado de siembras tumorales metastásicas y, por tanto, corresponde a enfermedad avanzada. Estas variantes deben ser conocidas por el clínico dado las connotaciones pronósticas y terapéuticas que ello conlleva.
Palabras clave: Tumores del estroma gastrointestinal. GIST. Tríada de Carney. Síndrome de Carney-Stratakis. Neurofibromatosis tipo I. Síndrome de GIST familiar. GIST pediátrico.
General features of gastrointestinal stromal tumors
Gastrointestinal stromal tumors (GISTs) are mesenchymal tumors of the gastrointestinal tract that differentiate towards interstitial cells of Cajal or their precursors; they are generally KIT (CD117)-positive, and display KIT or PDGFRA gene mutations (1,2). These tumors have recently prompted particular interest due to their good response to new targeted therapies (imatinib mesylate, Glivec®, Novartis) (3). They generally arise as solitary tumors of the stomach (50%), small intestine (25%), large intestine (10%) or esophagus (5%) (Fig. 1); in 10% of cases they occur outside the gastrointestinal tract (EGIST: extra-gastrointestinal stromal tumors), in the mesenterium, epiploon and -more rarely- appendix, gall bladder or pancreas (1,4). Epidemiological studies report an annual incidence of 6.8-19.6 per million inhabitants, depending on the countries surveyed (5), affecting men and women roughly equally, over a wide age range; however, 75% of cases occur in adults aged over 50 (6). The following clinical signs and symptoms may be observed: fatigue, abdominal pain, dysphagia, feeling of fullness, hematemesis or melena (1,4,5,7,8). Smaller GISTs are often incidental findings during surgery, radiological studies or endoscopy (1). At histological examination, tumors tend to display spindle cell, epithelioid or mixed cell morphology (Fig. 2). Immunohistochemical features include positivity for CD117 (95%), CD34 (60-70%), smooth muscle actin (30-40%), S-100 protein (5%), and more rarely desmin (1-2%) (1,4) (Fig. 3); it should be stressed that 5% of GISTs do not show positivity for CD117. Two new tissue markers (DOG-1, PKCθ) have been identified, providing specificity and sensitivity equal to or greater than those of CD117 (9-15). Both proteins are selectively expressed on interstitial cells of Cajal (ICC), but their relationship with the KIT receptor is unknown. Molecular studies have revealed mutually exclusive mutations in KIT (60-80%) and PGFRA (5-10%) genes (1,4,5,16). Recently, BRAF mutations were detected in 13% of cases bearing no KIT/PPDGFRA (wt KIT/PDGFRA) mutations (16). Approximately 20-25% of gastric and 40-50% of small intestinal GISTs are clinically malignant. Local recurrences and metastases (early and late) commonly develop in the abdominal cavity and liver; more rarely, in bone, soft tissue and skin (1). Lymph-node metastases are extremely rare, and primarily affect children (1). Tumor size, mitotic activity and location are the major prognostic factors (17-19).
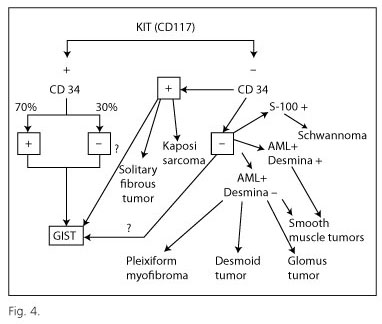
Diagnosis of GIST requires a basic immunohistochemical examination including the following markers: CD117, CD34, smooth muscle actin, desmin and S100 protein; these markers, routinely used in histology laboratories, enable differential diagnosis with respect to morphologically similar tumors (mainly leiomyoma/leiomyosarcoma and schwannoma) (Fig. 4). Molecular analysis is recommended for the diagnosis of CD117-negative tumors, and also as the basis for targeted therapy, since patients with GISTs harboring KIT exon 9 mutations need to double the dosage of imatinib (20), while those displaying the PDGRFA exon 18 mutation D842V fail to respond to this drug (21,22). In view of its specialized nature, molecular analysis is currently performed by reference centers.
Multiple gastrointestinal stromal tumors
In very rare cases, multiple GISTs may be detected in one or more organs; this is not necessarily an indicator of greater aggressivity. Digestive specialists need to be aware of these rare forms, whose prognosis and treatment differ from those of conventional GISTs. Multiple tumors may arise in three clinical contexts: sporadic tumor formation (in both pediatric and adult patients); familial GIST syndrome (autosomal dominant inheritance); or as an additional component of certain syndromes (Carney's triad, Carney-Stratakis syndrome and type I neurofibromatosis). The differential diagnosis of these syndromes is based mainly on clinical and genetic studies, rather than on morphological, immunohistochemical or molecular findings.
Multiple sporadic GISTs in adults
Multiple sporadic GISTs are generally characterized by the presence of two or three lesions, at the same site or in different sites, showing different molecular alterations in the same gene (KIT) (23,24), or in KIT and PDGFRA genes (25,26). Agaimy et al. (27) recently analyzed 11 patients aged over 70, with small sclerosing tumors or tumorlets (2-4 each) in the proximal stomach; tumors were incidental findings at autopsy or during surgery. All displayed spindle-cell morphology, and all were CD117+ and CD34+. In all but one case, individual lesions from the same patient displayed different KIT mutations (Table I). The presence of multiple sporadic lesions in the same patient points to the existence of distinct subsets of interstitial cells of Cajal or their precursors in different locations (field carcinogenesis theory); molecular analysis is therefore essential in order to distinguish metastatic from non-metastatic lesions (27).

Multiple sporadic GIST in children and adults under 30
GISTs in pediatric and adolescent patients account for 1-2% of all GISTs, and differ in certain respects from those recorded in adults (28). Molecular analysis has been performed in around 75 cases, and KIT/PDGFRA mutations have been detected in 11-15% of these (28-30). Pediatric GISTs are more prevalent in females (70%), occur prefentially in the stomach (88%), tend to be multifocal (81%), display epithelioid or mixed cell morphology (82%), and generally lack both KIT/PDGFRA mutations (88%) and interstitial cell of Cajal hyperplasia (100%). By contrast, the few pediatric GISTs with KIT or PDGFRA mutations have very different features: prevalence is greater in males, lesions tend to be unifocal, and usually occur in extragastric locations, and spindle-cell morphology is found in all cases (28,29); in other words, they share many of the features of spontaneous adult tumors.
Immunohistochemically, pediatric GISTs consistently overexpress CD117 (28-30) and DOG1 (10). Unlike in adults, pediatric GISTs tend to follow an indolent course, and are associated with better survival rates, despite the high rate of intra-abdominal and even lymph-node metastasis (28-30). The transcriptional signature is distinct from that of adult GISTs; the top-ranked genes overexpressed in the pediatric subset are CRLF1, BAALC, FGF4, PLAG1, and IGF1R. Tumor progression to malignancy is also different, and probably results from gene promoter methylation; unlike in adults, sequential chromosomal deletions (at 14q and 22q) and LOH are not common; by contrast, chromosomal additions (X, 1q, 5p, 8q, 9p, 12p, 13q, 18p, 19q) and amplifications (1q and 19p) are more frequent (28,29). The major differences between adult and pediatric subsets are shown in table II.

Although GIST is a heterogeneous entity in adults aged under 30, most cases display the predominance of mutation and the clinicoapathological features of the adult tumor (28).
Familial GIST syndrome
This is an extremely rare disorder. To date, a clear autosomal dominant inheritance pattern associated with one or more GISTs affecting two or more family members has been reported in only 20 families (31-49) (Table III). The phenotypic syndrome varies considerably, and may include -in addition to multiple, generally benign GISTs appearing in middle age- hyperpigmentation, urticaria pigmentosa and dysphagia (32,33). Germline mutations generally affect the KIT gene (31-46), though cases of PDGFRA germline mutations have also been reported (47-49). Skin alterations are common in patients with KIT exon 8 (31) and exon 11 mutations (32-36,39). Dysphagia, caused by esophageal dysmotility related to ICC hyperplasia, is observed in patients with KIT exon 8 (31) and exon 17 mutations (44,45).

KIT exon 17 mutations, which are very rare in sporadic GIST, have been reported in three cases of familial GIST; two at codon 820 (Asp820Tyr) (44,45) and one at codon 822 (Asn822Tyr) (46). The specific type of mutation is generally the same as that found in sporadic tumors, with two notable exceptions: KIT (exon 8) Asp419del (31) and PDGFRA (exon 12) Tyr555Cys (47). Pasini et al. (48) recently reported the unusual case of a patient carrying a germline PDGFRA mutation Asp561Val who developed multiple fibrous polyps (> 100) in the intestine, stomach, duodenum and cecum, lipomas in the duodenum and jejunum and gastric GIST, with no associated ICC hyperplasia. Gastric and duodenal fibrous tumors were CD34+, CD117-, PKCθ- and PDGFRA+; whilst the two gastric GISTs were CD117+, CD34+, PDGFRA+, PKθ+.
GIST associated with Carney's triad and the Carney-Stratakis syndrome
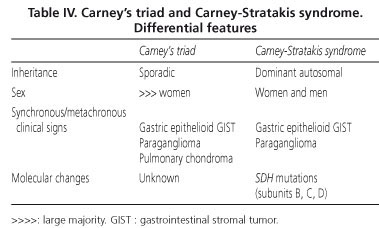
Carney's triad is a non-familial condition preferentially affecting women and combining the presence of multifocal epithelioid gastric GIST with the metachronous or synchronous occurrence of paraganglioma and pulmonary chondroma, although the conjunction of the three lesions is not necessary for diagnosis (50). Molecular studies to date have failed to reveal specific genetic alterations (51). Female predisposition, multifocal gastric location and wt KIT/PDGFRA genotype are findings which overlap considerably with clinicopathological features of pediatric GIST (28), suggesting that pediatric forms may represent a forme fruste of Carney's triad; longer follow-ups appear to confirm this suspicion, at least in some cases (28).
The Carney-Stratakis syndrome is a similar condition, in that it combines gastric GISTs and paragangliomas; however, it differs from Carney's triad in that it affects men and women with equal frequency, displays an autosomal dominant inheritance pattern and is not associated with pulmonary lesions; moreover, molecular analysis reveals germline mutations of the genes coding for SDH subunits B, C and D) (52,53). The main differential features of the two conditions are shown in table IV.
GIST associated with type I neurofibromatosis
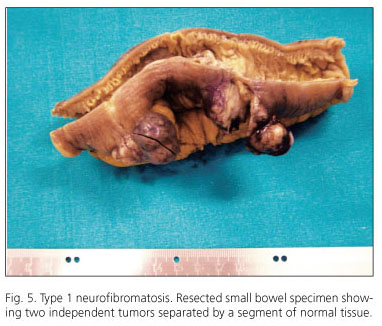
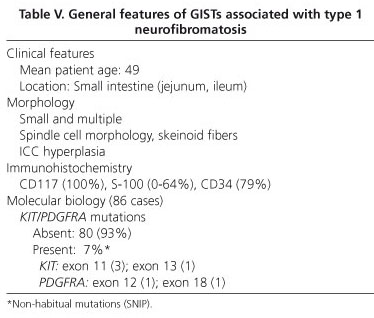
The range of gastrointestinal lesions associated with type 1 neurofibromatosis (NF1) includes: a) hyperplasia of nerve plexuses; b) GIST; c) endocrine tumors of the duodenum and periampullary region (somatostatinoma); and d) other tumors of varied histogenesis (54). The incidence of GIST in patients with NF1 is 7% (55). Most reported cases occur in middle-aged or elderly patients; tumors are multiple and often microscopic, are located in the small intestine (ileum and jejunum), and are accompanied by hyperplasia of the interstitial cells of Cajal (Fig. 5). Histologically, tumors display spindle-cell morphology, with few mitotic figures and frequent skeinoid fibers; they are immunohistochemically positive for CD117, DOG-1 and PKCθ (10,56-62) (Fig. 6). However, immunostaining for S100 protein yields variable results: some authors report low expression (58,61,62) or no expression at all (54,57) whilst others have observed positivity for S100 in up to 64% of cases (57). Molecular analysis in the seven largest cohorts indicates an incidence of KIT and PDGFRA mutations of 4.1% (6/145) and del 1.7% (2/116), respectively; some of these may be random mutations, since they are not observed in sporadic GISTs (57). A recent study has reported loss of heterozygosity at 14q (87.5%) and at 22q (41.7%), together with activation of the Ras-MAPK pathway, probably associated with inactivation of the NF1 gene (62). The main features of GISTs associated with type 1 neurofibromatosis are shown in table V.
References
1. Miettinen M, Lasota J. Gastrointestinal stromal tumors. Review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 2006; 130: 1466-78. [ Links ]
2. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998; 152: 1259-69. [ Links ]
3. Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001; 344: 1052-6. [ Links ]
4. Rubin BP. Gastrointestinal stromal tumors. An update. Histopathology 2006; 48: 83-96. [ Links ]
5. Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol Mech Dis 2008; 3: 557-86. [ Links ]
6. DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000; 231: 51-8. [ Links ]
7. Alberto S, Sánchez P, Oliveira M, Cuesta L, Gomes F, Figueiredo A, et al. Tumores del estroma gastrointestinal. Estudio retrospectivo de 43 casos. Rev Esp Enferm Dig 2008; 100: 696-700. [ Links ]
8. Fernández Salazar LI, Álvarez Gago T, Sanz Rubiales A, Velayos Jiménez B, Aller de la Fuente R, González Hernández JM. Tumores de la estroma gastrointestinal (GISTS): aspectos clínicos. Rev Esp Enferm Dig 2007; 99: 19-24. [ Links ]
9. Espinosa I, Lee CH, Kim MK, Rouse BT, Subramanian S, Montgomery K, et al. A novel monoclonal antibody against DOG1 is a sensitive and specific marker for gastrointestinal stromal tumors. Am J Surg Pathol 2008; 32: 210-8. [ Links ]
10. Liegl B, Hornick JL, Corless CL, Fletcher CD. Monoclonal antibody DOG1.1 shows higher sensitivity than kit in the diagnosis of gastrointestinal stromal tumors, including unusual subtypes Am J Surg Pathol 2009; 33: 437-46. [ Links ]
11. Miettinen M, Wang Z-F, Lasota J. DOG1 Antibody in the differential diagnosis of gastrointestinal stromal tumors A study of 1840 cases. Am J Surg Pathol 2009; 33: 1401-8. [ Links ]
12. Blay P, Astudillo A, Buesa JM, Campo E, Abad M, García-García J, et al. Protein kinase c is highly expressed in gastrointestinal stromal tumors but not in other mesenchymal neoplasias. Clin Cancer Res 2004; 10: 4089-95. [ Links ]
13. Duensing A, Joseph NE, Medeiros F, Smith F, Hornick JL, Heinrich MC, et al. Protein kinase C (PKC) expression and constitutive activation in gastrointestinal stromal tumors (GISTs). Cancer Res 2004; 64: 5127-31. [ Links ]
14. Lee HE, Kim MA, Lee HS, Lee BL, Kim WH. Characteristics of KIT-negative gastrointestinal stromal tumours and diagnostic utility of protein kinase C theta immunostaining. J Clin Pathol 2008; 61: 722-9. [ Links ]
15. Motegi A, Sakurai S, Nakayama H, Sano T, Oyama T, Nakajima T, et al. PKC theta, a novel immunohistochemical marker for gastrointestinal stromal tumors (GIST), especially useful for identifying KIT-negative tumors. Pathol Intern 2005; 55: 106-12. [ Links ]
16. Hostein I, Faur N, Primois C, Boury F, Denard J, Emile J-F, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol 2010; 133: 141-8. [ Links ]
17. Miettinen M, Sobin L, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immnuhistochemical and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol 2005; 29: 52-68. [ Links ]
18. Miettinen M, Maklhouf H, Sobin L, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileon: a clinicopathologic, immnuhistochemical and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol 2006; 30: 477-89. [ Links ]
19. Demetri GD, Benjamin RS, Blanke CD, Blay JY, Casali P, Choi H, et al. NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)-update of the NCCN Clinical Practice Guidelines. J Natl Compr Canc Netw 2007; 5(Supl. 2): S1-29. [ Links ]
20. Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006; 42: 1093-103. [ Links ]
21. Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003; 21: 4342-9. [ Links ]
22. Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005; 23: 5357-64. [ Links ]
23. Kang DY, Park CK, Choi JS, Jin SY, Kim HJ, Joo M, et al. Multiple gastrointestinal stromal tumors: clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol 2007; 31: 224-32. [ Links ]
24. Gasparotto D, Rossi S, Bearzi I, Doglioni C, Marzotto A, Hornick JL, et al. Multiple primary sporadic gastrointestinal stromal tumors in the adult: an underestimated entity. Clin Cancer Res 2008; 14: 5715-21. [ Links ]
25. Haller F, Schulten HJ, Armbrust T, Langer C, Gunawan B, Füzesi L. Multicentric sporadic gastrointestinal stromal tumors (GISTs) of the stomach with distinct clonal origin: differential diagnosis to familial and syndromal GIST variants and peritoneal metastasis. Am J Surg Pathol 2007; 31: 933-7. [ Links ]
26. Miselli F, Conca E, Casieri P, Grosso F, Schiavo M, Tamborini E, et al. A sporadic multiple GIST with unusual pathologic, molecular, and genetic features. Am J Surg Pathol 2008; 32: 340-1. [ Links ]
27. Agaimy A, Dirnhofer S, Wünsch PH, Terracciano LM, Tornillo L, Bihl MP. Multiple sporadic gastrointestinal stromal tumors (GISTs) of the proximal stomach are caused by different somatic KIT mutations suggesting a field effect. Am J Surg Pathol 2008; 32: 1553-9. [ Links ]
28. Agaram NP, Laquaglia MP, Ustun B, Guo T, Wong GC, Socci ND, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res 2008; 14: 3204-15. [ Links ]
29. Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, Corless CL, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res 2007; 67: 9084-8. [ Links ]
30. Miettinen M, Lasota J, Sobin LH. Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol 2005; 29: 1373-81. [ Links ]
31. Hartmann K, Wardelmann E, Ma Y, Merkelbach-Bruse S, Preussner LM, Woolery C, et al. Novel germline mutation of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology 2005; 129: 1042-6. [ Links ]
32. Robson ME, Glogowski E, Sommer G, Antonescu CR, Nafa K, Maki RG, et al. Pleomorphic characteristics of a germ-line KIT mutation in a large kindred with gastrointestinal stromal tumors, hyperpigmentation, and dysphagia. Clin Cancer Res 2004; 10: 1250-4. [ Links ]
33. Hirota S, Okazaki T, Kitamura Y, O'Brien P, Kapusta L, Dardick I. Cause of familial and multiple gastrointestinal autonomic nerve tumors with hyperplasia of interstitial cells of Cajal is germline mutation of the c-kit gene. Am J Surg Pathol 2000; 24: 326-7. [ Links ]
34. Beghini A, Tibiletti MG, Roversi G, Chiaravalli AM, Serio G, Capella C, et al. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer 2001; 92: 657-62. [ Links ]
35. Maeyama H, Hidaka E, Ota H, Minami S, Kajiyama M, Kuraishi A, et al. Familial gastrointestinal stromal tumor with hyperpigmentation: association with a germline mutation of the c-kit gene. Gastroenterology 2001; 120: 210-5. [ Links ]
36. Li FP, Fletcher JA, Heinrich MC, Garber JE, Sallan SE, Curiel-Lewandrowski C, et al. Familial gastrointestinal stromal tumor syndrome: phenotypic and molecular features in a kindred. J Clin Oncol 2005; 23: 2735-43. [ Links ]
37. Kang DY, Park CK, Choi JS, Jin SY, Kim HJ, Joo M, et al. Multiple gastrointestinal stromal tumors: clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol 2007; 31: 224-32. [ Links ]
38. Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 1998; 19: 323-4. [ Links ]
39. Carballo M, Roig I, Aguilar F, Pol MA, Gamundi MJ, Hernan I, et al. Novel c-KIT germline mutation in a family with gastrointestinal stromal tumors and cutaneous hyperpigmantation. Am J Med Genet A 2005; 132: 361-4. [ Links ]
40. Lasota J, Miettinen M. A new familial GIST identified. Am J Surg Pathol 2006; 30: 1342. [ Links ]
41. Tarn C, Merkel E, Canutescu AA, Shen W, Skorobogatko Y, Heslin MJ, et al. Analysis of KIT mutations in sporadic ad familial gastrointestinal stromal tumors: therapeutic implications through protein modeling. Clin Cancer Res 2005; 11: 3668-77. [ Links ]
42. Isozaki K, Terris B, Belghiti J, Schiffmann S, Hirota S, Vanderwinden JM. Germline-activating mutation in the kinase domain on KIT gene in familial gastrointestinal stromal tumors. Am J Pathol 2001; 157: 1581-5. [ Links ]
43. Graham J, Debiec-Rychter M, Corless CL, Reid R, Davidson R, White JD. Imatinib in the management of multiple gastrointestinal stromal tumors associated with a germline KIT K642E mutation. Arch Pathol Lab Med 2007; 131: 1393-6. [ Links ]
44. Hirota S, Nishida T, Isozaki K, Taniguchi M, Nishikawa K, Ohashi A, et al. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology 2002; 122: 1493-9. [ Links ]
45. O'Riain C, Corless CL, Heinrich MC, Keegan D, Vioreanu M, Maguire D, et al. Gastrointestinal stromal tumors: insights from a new familial GIST kindred with unusual genetic and pathologic features. Am J Surg Pathol 2005; 29: 1680-3. [ Links ]
46. Thalheimer A, Schlemmer M, Bueter M, Merkelbach-Bruse S, Schildhaus H-U, Buettner R, et al. Familial gastrointestinal stromal tumors caused by the novel KIT exon 17 germline mutation N822Y. Am J Surg Pathol 2008; 32: 1560-5. [ Links ]
47. de Raedt T, Cools J, Debiec-Rychter M, Brems H, Mentens N, Sciot R, et al. Intestinal neurofibromatosis is a subtype of familial GIST and results from dominant activating mutation in PDGFRA. Gastroenterology 2006; 131: 1907-12. [ Links ]
48. Pasini B, Matyakhina L, Bei T, Muchow M, Boikos S, Ferrando B, et al. Multiple gastrointestinal stromal tumors caused by platelet-derived growth factor receptor a gene mutations: a case associated with a germline V561D defect. J Clin Endocrin Metab 2007; 92: 3728-32. [ Links ]
49. Chompret A, Kannengiesser C, Barrois M, Terrier P, Dahan P, Tursz T, et al. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004; 126: 318-21. [ Links ]
50. Carney JA, Sheps SG, Go VL, Gordon H. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med 1977; 296: 1517-8. [ Links ]
51. Agaimy A, Pelz AF, Corless CL, Wünsch PH, Heinrich MC, Hofstaedter F, et al. Epithelioid gastric stromal tumours of the antrum in young females with the Carney triad: a report of three new cases with mutational analysis and comparative genomic hybridization. Oncol Rep 2007; 18: 9-15. [ Links ]
52. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet 2002; 108: 132-9. [ Links ]
53. Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 2008; 16: 79-88. [ Links ]
54. Fuller CE, Williams GT. Gastrointestinal manifestations of type 1 neurofibromatosis (von Recklinghausen's disease). Histopathology 1991; 19: 1-11. [ Links ]
55. Zöller ME, Rembeck B, Odén A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer 1997; 79: 2125-31. [ Links ]
56. Andersson J, Sihto H, Meis-Kindblom JM, Joensuu H, Nupponen N, Kindblom LG. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol 2005; 29: 1170-6. [ Links ]
57. Takazawa Y, Sakurai S, Sakuma Y, Ikeda T, Yamaguchi J, Hashizume Y, et al. Gastrointestinal stromal tumors of neurofibromatosis type 1 (von Rcklinghausen's disease). Am J Surg Pathol 2005; 29: 755-63. [ Links ]
58. Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol 2006; 30: 90-6. [ Links ]
59. Yantiss RK, Rosenberg AE, Sarran L, Besmer P, Antonescu CR. Multiple gastrointestinal stromal tumors in type I neurofibromatosis: a pathologic and molecular study. Mod Pathol 2005; 18: 475-84. [ Links ]
60. Kinoshita K, Hirota S, Isozaki K, Ohashi A, Nishida T, Kitamura Y, et al. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol 2004; 202: 80-5. [ Links ]
61. Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 2006; 15: 1015-23. [ Links ]
62. Yamamoto H, Tobo T, Nakamori M, Imamura M, Kojima A, Oda Y, et al. Neurofibromatosis type 1-related gastrointestinal stromal tumors: a special reference to loss of heterozygosity at 14q and 22q. J Cancer Res Clin Oncol 2009; 135: 791-8. [ Links ]
![]() Correspondence:
Correspondence:
Ricardo González Cámpora.
Servicio de Anatomía Patológica.
Hospital Universitario Virgen Macarena.
Avda. Dr. Fedriani, 3.
41009 Sevilla, Spain.
e-mail: rcampora@us.es
Received: 18-01-10.
Accepted: 18-03-10.

