










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versão impressa ISSN 1130-0108
Rev. esp. enferm. dig. vol.102 no.8 Madrid Ago. 2010
Tumores del estroma gastrointestinal múltiples no metastásicos. Aspectos diferenciales
Multiple non-metastatic gastrointestinal stromal tumors. Differential features
M. Díaz-Delgado1, A. Hernández-Amate2, M. Sánchez-León3, S. Pereira-Gallardo3, E. Prieto-Sánchez4, M. Jiménez-Sáenz5 y R. González-Cámpora3
Servicio de Anatomía Patológica. 1Hospital de Mérida. Mérida, Badajoz. 2Hospital Universitario Infanta Cristina. Badajoz. 3Hospital Universitario Virgen Macarena. Sevilla. 4Hospital de Motril. Granada. 5Servicio de Aparato Digestivo. Hospital Universitario Virgen Macarena. Sevilla
Dirección para correspondencia
RESUMEN
Introducción: los tumores del estroma gastrointestinal (GIST) son neoplasias mesenquimales del tubo digestivo que generalmente expresan el receptor KIT (CD117) y muestran mutaciones en los genes KIT o PDGFRA. Aunque la forma de presentación clínica habitual es como una neoplasia mural solitaria, excepcionalmente pueden presentarse formas múltiples en el mismo o diferente órgano.
Objetivo: revisar las características morfológicas, inmunohistoquímicas y moleculares de las formas de GIST múltiples no metastásicos.
Fuentes: revisión de la literatura en Medline y la propia experiencia.
Conclusiones: los GIST múltiples pueden presentarse en tres contextos diferentes: lesiones espontáneas (del adulto o de la edad infantil); síndrome familiar propio (transmitido con herencia autosómica dominante); y lesiones asociadas a síndromes específicos (tríada de Carney, síndrome de Carney-Stratakis, y neurofibromatosis tipo I). Fuera de estos ámbitos, se interpreta que todo GIST múltiple es el resultado de siembras tumorales metastásicas y, por tanto, corresponde a enfermedad avanzada. Estas variantes deben ser conocidas por el clínico dado las connotaciones pronósticas y terapéuticas que ello conlleva.
Palabras clave: Tumores del estroma gastrointestinal. GIST. Tríada de Carney. Síndrome de Carney-Stratakis. Neurofibromatosis tipo I. Síndrome de GIST familiar. GIST pediátrico.
ABSTRACT
Introduction: gastrointestinal stromal tumors (GISTs) are specific, generally KIT (CD117)-positive, mesenchymal tumors of the digestive tract displaying KIT or PDGFRA gene mutations. Clinically, they tend to present as solitary tumors of the intestinal wall; more rarely, multiple tumors may occur in one or more organs.
Objective: to review the morphological, immunohistochemical and molecular features of multiple, non-metastatic forms of GIST.
Sources: review of the literature on Medline, and authors' own experience.
Conclusions: multiples GISTs may occur in three different contexts: as spontaneous lesions (in both adults and children); due to familial GIST syndrome (autosomal dominant inheritance); or in association with specific syndromes (e.g. Carney's triad, Carney-Stratakis syndrome, type I neurofibromatosis). Outside these contexts, the existence of multiple GISTs is deemed to be the result of tumor metastasis, and therefore indicative of advanced-stage disease. Clinicians need to be aware of these variants, whose prognosis and treatment differ.
Key words: Gastrointestinal stromal tumors. GIST. Carney's triad. Carney-Stratakis syndrome. Type I neurofibromatosis. Familial GIST syndrome. Pediatric GIST.
Características generales de los tumores del estroma gastrointestinal
Los tumores del estroma gastrointestinal (GIST) son neoplasias mesenquimales específicas del tubo gastrointestinal que muestran diferenciación hacia las células intersticiales de Cajal o a sus precursores; generalmente expresan el receptor KIT (CD117) y presentan mutaciones en los genes KIT o PDGFRA (1,2). El interés especial que existe en la actualidad por estos tumores reside en su buena respuesta al tratamiento con las nuevas terapias dirigidas (mesilato de imatinib, Glivec®, Novartis) (3). Generalmente se presentan como lesiones solitarias en el estómago (50%), intestino delgado (25%), intestino grueso (10%) y esófago (5%) (Fig. 1); en el 10% de los casos asientan fuera del tubo digestivo (GIST extra-gastrointestinales) en mesenterio, epiplón y, más raramente en apéndice, vesícula y páncreas (1,4). Los estudios epidemiológicos han revelado una incidencia anual de 6,8-19,6 por millón de habitantes en función de los países analizados (5), sin diferencias en género y amplia distribución etaria; no obstante, el 75% de los casos se observan en adultos mayores de 50 años (6). Clínicamente pueden presentarse con los siguientes signos y síntomas: fatiga, dolor abdominal, disfagia, sensación de saciedad, hematemesis o melenas (1,4,5,7,8). Las lesiones más pequeñas generalmente son hallazgos casuales endoscópicos, radiológicos o quirúrgicos (1). Histológicamente pueden mostrar una citomorfología fusocelular, epiteliode o mixta (Fig. 2) con el siguiente perfil inmunohistoquímico: CD117 (95%), CD34 (60-70%), actina de músculo liso (30-40%), proteína S-100(5%), desmina (1-2%) (1,4) (Fig. 3); es de destacar que el 5% de los GIST no expresan inmunorreacción positiva al CD117. Recientemente se han introducido dos nuevos marcadores tisulares (DOG-1, PKCθ) que presentan mayor o igual especificidad y sensibilidad que el CD117 (9-15). Ambas proteínas se expresan selectivamente en las células intersticiales de Cajal pero se desconoce su relación con el receptor KIT. Los estudios moleculares han revelado la existencia de mutaciones excluyentes en los en los genes KIT (60-80%) y PGFRA (5-10%) (1,4,5,16). Recientemente, en el 13% casos sin mutaciones en KIT/PPDGFRA (wt KIT/PDGFRA) se han encontrado mutaciones en el gen BRAF (16). Aproximadamente el 20-25% de los casos gástricos y el 40-50% de los del intestino delgado tienen un curso clínico maligno, con recidivas locales y metástasis (precoces o tardías) en la cavidad abdominal e hígado; más raramente también pueden asentar en hueso, tejidos blandos y piel (1). Las metástasis en ganglios linfáticos son excepcionales y se presentan preferentemente en la población infantil (1). Los factores que mejor se correlacionan con la evolución son el tamaño tumoral, índice de mitosis y localización (17-19).
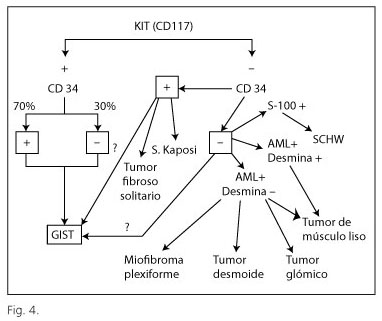
Para poder realizar el diagnóstico de GIST es requisito necesario llevar a cabo un estudio inmunohistoquímico básico que comprende los siguientes marcadores: CD117, CD34, actina de músculo liso, desmina y proteína S-100; estos marcadores son de uso habitual en los laboratorios de Anatomía Patológica y permiten establecer el diagnóstico diferencial con lesiones morfológicamente semejantes (principalmente leiomioma/leiomiosarcoma, schwannoma) (Fig. 4). El estudio molecular es recomendable en los tumores CD117-negativos con fines diagnósticos pero también son de utilidad para dirigir la terapia, puesto que los pacientes con tumores con mutaciones en el exón 9 del KIT necesitan duplicar la dosis de Glivec® (20) y los portadores de la mutación D842V en el exón 18 del gen PDGFRA no presentan ningún tipo de respuesta (21,22). Los análisis moleculares, debido a su carácter especializado, se llevan a cabo por ahora en centros de referencia.
Tumores del estroma gastrointestinal múltiples
Excepcionalmente, los GIST pueden presentarse como lesiones múltiples en el mismo o diferente órgano sin que ello comporte mayor grado de agresividad. El especialista del aparato digestivo debe tener conocimiento de estas formas especiales dado que conllevan connotaciones pronósticas y terapéuticas diferentes a los GIST convencionales. Estas variedades peculiares de GIST pueden presentarse en tres contextos clínicos diferentes: tumoraciones esporádicas (tanto en el adulto como en la edad pediátrica), formando un síndrome familiar propio (herencia autosómica dominante) y como componente adicional de ciertos síndromes (tríada de Carney, síndrome de Carney-Stratakis y neurofibromatosis tipo I). El diagnóstico diferencial entre estos cuadros descansa principalmente en el estudio clínico y genético, no en detalles morfológicos, inmunohistoquímicos o moleculares.
GIST múltiples esporádicos del adulto
Se caracterizan por ser dos o tres lesiones, generalmente sincrónicas, que asientan en el mismo o en diferente órgano y que muestran alteraciones moleculares diferentes en el mismo gen (KIT) (23,24), o en los genes KIT y PDGFRA (25,26). Recientemente, Agaimy y cols. (27) han descrito 11 pacientes, con edades superiores a 70 años, que presentaban tumores múltiples (2 a 4), pequeños y esclerosantes (tumorlets), en la parte proximal del estómago, que fueron hallazgos incidentales en autopsia o intervenciones quirúrgicas. Todos ellos estaban constituidos por células fusiformes, expresaban CD117 y CD34 + y en todos, excepto uno, presentaban mutaciones diferentes en el gen KIT en las distintas neoplasias (Tabla I). La existencia de lesiones múltiples esporádicas abre la posibilidad de desarrollo lesional a partir de distintas células intersticiales de Cajal o de sus precursores en diferentes localizaciones (teoría de la carcinogénesis de campo) y obliga a realizar estudio molecular antes de indicar que determinadas lesiones corresponden a siembras metastásicas (27).

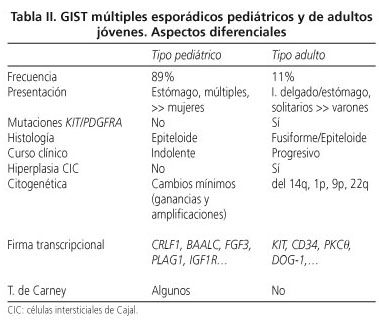
GIST múltiples esporádicos pediátricos y de adultos jóvenes (< 30 años)
Los GIST en la edad pediátrica y de la adolescencia representan el 1-2% del total de GIST y muestran ciertas características diferenciales en relación con los del adulto (28). Aproximadamente se han descrito 75 casos con estudio molecular, encontrándose mutaciones en KIT/PDGFRA en el 11-15% de los casos (28-30). Estas lesiones se caracterizan por afectar preferentemente a mujeres (70%), localizarse en el estómago (88%), ser multifocales (81%), presentar celularidad epitelioide o mixta (82%), y carecer de mutaciones en KIT/PDGFRA (88%) y de hiperplasia de células intersticiales de Cajal (100%). Los escasos GIST pediátricos con mutaciones en KIT o PDGFRA, en cambio, tienen unas características totalmente diferentes: afectan preferentemente a varones, suelen ser unifocales, de localización extragástrica y todos ellos presentan celularidad fusiforme (28,29); es decir, muestran muchas de las características propias de los tumores espontáneos del adulto.
Con las técnicas de inmunohistoquímica los GIST pediátricos expresan fuertemente CD117 (28-30) y DOG1 (10) y el curso clínico es más indolente que en el adulto, con supervivencias más prolongadas, a pesar de presentar metástasis intraabdominales e incluso ganglionares (28-30). El perfil transcripcional y la progresión tumoral en los GIST pediátricos son también totalmente diferentes a los del adulto, ya que existe sobreexpresión de CRLF1, BAALC, FGF4, PLAG1, IGF1R. La progresión tumoral probablemente se realiza mediante metilaciones y pérdidas cromosómicas (del 14q y 22q), no siendo habituales las LOH, como sucede en el adulto; en cambio, son más frecuentes las ganancias cromosómicas (X, 1q, 5p, 8q, 9p, 12p, 13q, 18p, 19q) y amplificaciones (1q y 19p) (28,29). La tabla II recoge las diferencias fundamentales entre estos dos grupos tumorales.
En los adultos < 30 años, los GIST también constituyen un grupo heterogéneo pero con predominio de formas mutadas y características clinicopatológicas propias de los tumores del adulto (28).
Síndrome del GIST familiar
Es un trastorno muy poco común. Hasta la fecha se han descrito 20 familias con un claro patrón de herencia autosómica dominante que cursen con uno o varios GIST en dos o más miembros de la familia (31-49) (Tabla III). El cuadro fenotípico es muy variable y puede incluir -además de múltiples GIST de aparición en edad media y generalmente benignos- hiperpigmentación, urticaria pigmentosa y disfagia (32,33). En la mayoría de las ocasiones las mutaciones en línea germinal se presentan en el gen KIT (31-46), pero también se han señalado casos con mutaciones en PDGFRA (47-49). Las alteraciones cutáneas son comunes en pacientes con mutaciones en los exones 8 (31) y 11 del KIT (32-36,39). La disfagia, causada por dismotilidad esofágica y relacionada con hiperplasia de células intersticiales de Cajal se observa en pacientes con mutaciones en los exones 8 (31) y 17 (44,45) del gen KIT.
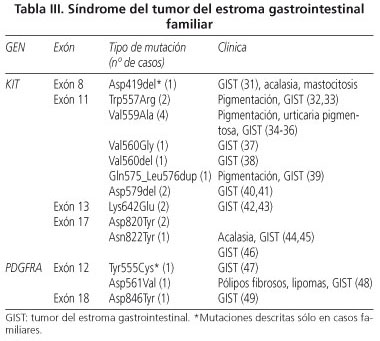
Las mutaciones en el exón 17 del KIT, que son muy raras en los GIST esporádicos, se han descrito en tres casos familiares; dos en el codón 820 (Asp820Tyr) (44,45) y una en el 822 (Asn822Tyr) (46). Es de señalar que el tipo específico de mutaciones por lo general es el mismo que el que se encuentra en los tumores esporádicos, salvo dos excepciones: KIT (exón 8) Asp419del (31) y PDGFRA (exón 12) Tyr555Cys (47). Recientemente Pasini y cols. (48) han descrito un caso singular con mutaciones en PDGFRA Asp561Val que desarrolló múltiples pólipos fibrosos (> 100) en intestino, estómago, duodeno y apéndice cecal, lipomas en duodeno y yeyuno y GIST gástrico, en ausencia de hiperplasia de células de Cajal asociada. Los tumores fibrosos gástricos y duodenales fueron CD34+, CD117-, PKCθ- y PDGFRA+; mientras que los dos GIST gástricos fueron CD117+, CD34+, PDGFRA+, PKθ+.
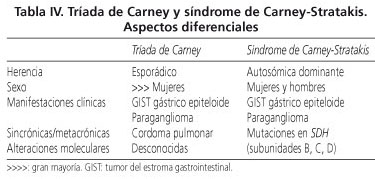
GIST asociados a la tríada de Carney y síndrome de Carney-Stratakis
La tríada de Carney es un trastorno no familiar, caracterizado por afectar preferentemente a mujeres y combinar la presencia de un GIST gástrico epiteloide multicéntrico con la aparición, sincrónica o metacrónica, de paraganglioma y de condroma pulmonar, aunque no es necesaria la existencia de las tres lesiones para realizar el diagnóstico (50). Los estudios moleculares hasta la fecha no han revelado alteraciones específicas (51). El predominio por el sexo femenino, la multicentricidad, preferencia por el estómago y genotipo wt KIT/PDGFRA son hallazgos totalmente superponibles a los encontrados en la mayoría de los casos pediátricos (28), por lo que probablemente las formas pediátricas corresponden a formas frustras de la tríada de Carney; de hecho, los seguimientos prolongados parecen confirmar esta sospecha al menos en algunos casos (28).
El síndrome de Carney-Stratakis es un cuadro similar donde se combinan GIST gástricos y paragangliomas pero, a diferencia con la tríada de Carney, afecta por igual a hombres y mujeres, muestra un patrón de herencia autosómico dominante, y carece de lesiones pulmonares; además, en el estudio molecular existen mutaciones en el gen SDH (subunidades B, C y D) (52,53). La tabla IV recoge las características diferenciales más significativas de estas dos entidades.
GIST asociados a neurofibromatosis tipo I
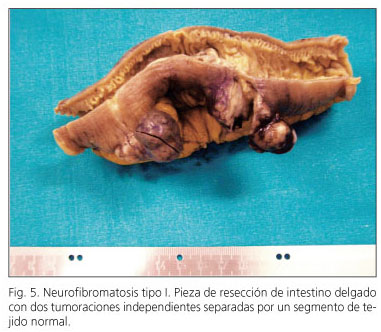
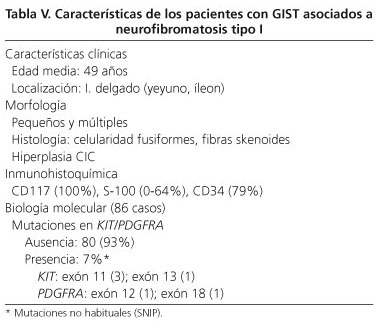
El espectro de lesiones gastrointestinales asociadas a neurofibromatosis tipo 1 incluye: a) hiperplasia de plexos nerviosos; b) GIST; c) tumores endocrinos del duodeno y región periampular (somatostatinoma); y d) otros tumores de histogénesis variada (54). La incidencia de GIST en pacientes con neurofibromatosis tipo 1 es del 7% (55). La mayoría de los casos descritos se presentan en adultos de edad media-avanzada, son múltiples y con frecuencia microscópicos, se localizan en intestino delgado (íleon o yeyuno) y se acompañan de hiperplasia de las células intersticiales de Cajal (Fig. 5). Histológicamente tienen celularidad fusiforme, con muy escasas figuras de mitosis, frecuentes fibras skenoides e inmunorreacción positiva al CD117, DOG-1 y PKCθ (10, 56-62) (Fig. 6). Los resultados con la inmunotinción a la proteína S-100, en cambio, son muy variables, mientras que algunos refieren ausencia de expresión (54,57) o expresión baja (58,61,62), otros señalan positividad hasta en el 64% del los casos (57). La incidencia de mutaciones en KIT y PDGFRA, de acuerdo con las siete series mayores, es del 4,1% (6/145) y del 1,7% (2/116), respectivamente, siendo de señalar que alguna de estas podrían considerarse como mutaciones al azar, ya que no están descritas en GIST esporádicos (57). Recientemente se ha señalado pérdida de heterozigosidad en 14q (87,5%) y en 22q (41,7%) y activación de la vía RAS-MAPK, probablemente relacionada con la inactivación del NF1 (62). En la tabla V se resumen las características principales de los GIST asociados a neurofibromatosis tipo I.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Ricardo González Cámpora.
Servicio de Anatomía Patológica.
Hospital Universitario Virgen Macarena.
Avda. Dr. Fedriani, 3.
41009 Sevilla.
e-mail: rcampora@us.es
Recibido: 18-01-10.
Aceptado: 18-03-10.
Bibliografía
1. Miettinen M, Lasota J. Gastrointestinal stromal tumors. Review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 2006; 130: 1466-78. [ Links ]
2. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998; 152: 1259-69. [ Links ]
3. Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001; 344: 1052-6. [ Links ]
4. Rubin BP. Gastrointestinal stromal tumors. An update. Histopathology 2006; 48: 83-96. [ Links ]
5. Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol Mech Dis 2008; 3: 557-86. [ Links ]
6. DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000; 231: 51-8. [ Links ]
7. Alberto S, Sánchez P, Oliveira M, Cuesta L, Gomes F, Figueiredo A, et al. Tumores del estroma gastrointestinal. Estudio retrospectivo de 43 casos. Rev Esp Enferm Dig 2008; 100: 696-700. [ Links ]
8. Fernández Salazar LI, Álvarez Gago T, Sanz Rubiales A, Velayos Jiménez B, Aller de la Fuente R, González Hernández JM. Tumores de la estroma gastrointestinal (GISTS): aspectos clínicos. Rev Esp Enferm Dig 2007; 99: 19-24. [ Links ]
9. Espinosa I, Lee CH, Kim MK, Rouse BT, Subramanian S, Montgomery K, et al. A novel monoclonal antibody against DOG1 is a sensitive and specific marker for gastrointestinal stromal tumors. Am J Surg Pathol 2008; 32: 210-8. [ Links ]
10. Liegl B, Hornick JL, Corless CL, Fletcher CD. Monoclonal antibody DOG1.1 shows higher sensitivity than kit in the diagnosis of gastrointestinal stromal tumors, including unusual subtypes Am J Surg Pathol 2009; 33: 437-46. [ Links ]
11. Miettinen M, Wang Z-F, Lasota J. DOG1 Antibody in the differential diagnosis of gastrointestinal stromal tumors A study of 1840 cases. Am J Surg Pathol 2009; 33: 1401-8. [ Links ]
12. Blay P, Astudillo A, Buesa JM, Campo E, Abad M, García-García J, et al. Protein kinase c is highly expressed in gastrointestinal stromal tumors but not in other mesenchymal neoplasias. Clin Cancer Res 2004; 10: 4089-95. [ Links ]
13. Duensing A, Joseph NE, Medeiros F, Smith F, Hornick JL, Heinrich MC, et al. Protein kinase C (PKC) expression and constitutive activation in gastrointestinal stromal tumors (GISTs). Cancer Res 2004; 64: 5127-31. [ Links ]
14. Lee HE, Kim MA, Lee HS, Lee BL, Kim WH. Characteristics of KIT-negative gastrointestinal stromal tumours and diagnostic utility of protein kinase C theta immunostaining. J Clin Pathol 2008; 61: 722-9. [ Links ]
15. Motegi A, Sakurai S, Nakayama H, Sano T, Oyama T, Nakajima T, et al. PKC theta, a novel immunohistochemical marker for gastrointestinal stromal tumors (GIST), especially useful for identifying KIT-negative tumors. Pathol Intern 2005; 55: 106-12. [ Links ]
16. Hostein I, Faur N, Primois C, Boury F, Denard J, Emile J-F, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol 2010; 133: 141-8. [ Links ]
17. Miettinen M, Sobin L, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immnuhistochemical and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol 2005; 29: 52-68. [ Links ]
18. Miettinen M, Maklhouf H, Sobin L, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileon: a clinicopathologic, immnuhistochemical and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol 2006; 30: 477-89. [ Links ]
19. Demetri GD, Benjamin RS, Blanke CD, Blay JY, Casali P, Choi H, et al. NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)-update of the NCCN Clinical Practice Guidelines. J Natl Compr Canc Netw 2007; 5(Supl. 2): S1-29. [ Links ]
20. Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006; 42: 1093-103. [ Links ]
21. Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003; 21: 4342-9. [ Links ]
22. Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005; 23: 5357-64. [ Links ]
23. Kang DY, Park CK, Choi JS, Jin SY, Kim HJ, Joo M, et al. Multiple gastrointestinal stromal tumors: clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol 2007; 31: 224-32. [ Links ]
24. Gasparotto D, Rossi S, Bearzi I, Doglioni C, Marzotto A, Hornick JL, et al. Multiple primary sporadic gastrointestinal stromal tumors in the adult: an underestimated entity. Clin Cancer Res 2008; 14: 5715-21. [ Links ]
25. Haller F, Schulten HJ, Armbrust T, Langer C, Gunawan B, Füzesi L. Multicentric sporadic gastrointestinal stromal tumors (GISTs) of the stomach with distinct clonal origin: differential diagnosis to familial and syndromal GIST variants and peritoneal metastasis. Am J Surg Pathol 2007; 31: 933-7. [ Links ]
26. Miselli F, Conca E, Casieri P, Grosso F, Schiavo M, Tamborini E, et al. A sporadic multiple GIST with unusual pathologic, molecular, and genetic features. Am J Surg Pathol 2008; 32: 340-1. [ Links ]
27. Agaimy A, Dirnhofer S, Wünsch PH, Terracciano LM, Tornillo L, Bihl MP. Multiple sporadic gastrointestinal stromal tumors (GISTs) of the proximal stomach are caused by different somatic KIT mutations suggesting a field effect. Am J Surg Pathol 2008; 32: 1553-9. [ Links ]
28. Agaram NP, Laquaglia MP, Ustun B, Guo T, Wong GC, Socci ND, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res 2008; 14: 3204-15. [ Links ]
29. Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, Corless CL, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res 2007; 67: 9084-8. [ Links ]
30. Miettinen M, Lasota J, Sobin LH. Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol 2005; 29: 1373-81. [ Links ]
31. Hartmann K, Wardelmann E, Ma Y, Merkelbach-Bruse S, Preussner LM, Woolery C, et al. Novel germline mutation of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology 2005; 129: 1042-6. [ Links ]
32. Robson ME, Glogowski E, Sommer G, Antonescu CR, Nafa K, Maki RG, et al. Pleomorphic characteristics of a germ-line KIT mutation in a large kindred with gastrointestinal stromal tumors, hyperpigmentation, and dysphagia. Clin Cancer Res 2004; 10: 1250-4. [ Links ]
33. Hirota S, Okazaki T, Kitamura Y, O'Brien P, Kapusta L, Dardick I. Cause of familial and multiple gastrointestinal autonomic nerve tumors with hyperplasia of interstitial cells of Cajal is germline mutation of the c-kit gene. Am J Surg Pathol 2000; 24: 326-7. [ Links ]
34. Beghini A, Tibiletti MG, Roversi G, Chiaravalli AM, Serio G, Capella C, et al. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer 2001; 92: 657-62. [ Links ]
35. Maeyama H, Hidaka E, Ota H, Minami S, Kajiyama M, Kuraishi A, et al. Familial gastrointestinal stromal tumor with hyperpigmentation: association with a germline mutation of the c-kit gene. Gastroenterology 2001; 120: 210-5. [ Links ]
36. Li FP, Fletcher JA, Heinrich MC, Garber JE, Sallan SE, Curiel-Lewandrowski C, et al. Familial gastrointestinal stromal tumor syndrome: phenotypic and molecular features in a kindred. J Clin Oncol 2005; 23: 2735-43. [ Links ]
37. Kang DY, Park CK, Choi JS, Jin SY, Kim HJ, Joo M, et al. Multiple gastrointestinal stromal tumors: clinicopathologic and genetic analysis of 12 patients. Am J Surg Pathol 2007; 31: 224-32. [ Links ]
38. Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 1998; 19: 323-4. [ Links ]
39. Carballo M, Roig I, Aguilar F, Pol MA, Gamundi MJ, Hernan I, et al. Novel c-KIT germline mutation in a family with gastrointestinal stromal tumors and cutaneous hyperpigmantation. Am J Med Genet A 2005; 132: 361-4. [ Links ]
40. Lasota J, Miettinen M. A new familial GIST identified. Am J Surg Pathol 2006; 30: 1342. [ Links ]
41. Tarn C, Merkel E, Canutescu AA, Shen W, Skorobogatko Y, Heslin MJ, et al. Analysis of KIT mutations in sporadic ad familial gastrointestinal stromal tumors: therapeutic implications through protein modeling. Clin Cancer Res 2005; 11: 3668-77. [ Links ]
42. Isozaki K, Terris B, Belghiti J, Schiffmann S, Hirota S, Vanderwinden JM. Germline-activating mutation in the kinase domain on KIT gene in familial gastrointestinal stromal tumors. Am J Pathol 2001; 157: 1581-5. [ Links ]
43. Graham J, Debiec-Rychter M, Corless CL, Reid R, Davidson R, White JD. Imatinib in the management of multiple gastrointestinal stromal tumors associated with a germline KIT K642E mutation. Arch Pathol Lab Med 2007; 131: 1393-6. [ Links ]
44. Hirota S, Nishida T, Isozaki K, Taniguchi M, Nishikawa K, Ohashi A, et al. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology 2002; 122: 1493-9. [ Links ]
45. O'Riain C, Corless CL, Heinrich MC, Keegan D, Vioreanu M, Maguire D, et al. Gastrointestinal stromal tumors: insights from a new familial GIST kindred with unusual genetic and pathologic features. Am J Surg Pathol 2005; 29: 1680-3. [ Links ]
46. Thalheimer A, Schlemmer M, Bueter M, Merkelbach-Bruse S, Schildhaus H-U, Buettner R, et al. Familial gastrointestinal stromal tumors caused by the novel KIT exon 17 germline mutation N822Y. Am J Surg Pathol 2008; 32: 1560-5. [ Links ]
47. de Raedt T, Cools J, Debiec-Rychter M, Brems H, Mentens N, Sciot R, et al. Intestinal neurofibromatosis is a subtype of familial GIST and results from dominant activating mutation in PDGFRA. Gastroenterology 2006; 131: 1907-12. [ Links ]
48. Pasini B, Matyakhina L, Bei T, Muchow M, Boikos S, Ferrando B, et al. Multiple gastrointestinal stromal tumors caused by platelet-derived growth factor receptor a gene mutations: a case associated with a germline V561D defect. J Clin Endocrin Metab 2007; 92: 3728-32. [ Links ]
49. Chompret A, Kannengiesser C, Barrois M, Terrier P, Dahan P, Tursz T, et al. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004; 126: 318-21. [ Links ]
50. Carney JA, Sheps SG, Go VL, Gordon H. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med 1977; 296: 1517-8. [ Links ]
51. Agaimy A, Pelz AF, Corless CL, Wünsch PH, Heinrich MC, Hofstaedter F, et al. Epithelioid gastric stromal tumours of the antrum in young females with the Carney triad: a report of three new cases with mutational analysis and comparative genomic hybridization. Oncol Rep 2007; 18: 9-15. [ Links ]
52. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet 2002; 108: 132-9. [ Links ]
53. Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 2008; 16: 79-88. [ Links ]
54. Fuller CE, Williams GT. Gastrointestinal manifestations of type 1 neurofibromatosis (von Recklinghausen's disease). Histopathology 1991; 19: 1-11. [ Links ]
55. Zöller ME, Rembeck B, Odén A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer 1997; 79: 2125-31. [ Links ]
56. Andersson J, Sihto H, Meis-Kindblom JM, Joensuu H, Nupponen N, Kindblom LG. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol 2005; 29: 1170-6. [ Links ]
57. Takazawa Y, Sakurai S, Sakuma Y, Ikeda T, Yamaguchi J, Hashizume Y, et al. Gastrointestinal stromal tumors of neurofibromatosis type 1 (von Rcklinghausen's disease). Am J Surg Pathol 2005; 29: 755-63. [ Links ]
58. Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol 2006; 30: 90-6. [ Links ]
59. Yantiss RK, Rosenberg AE, Sarran L, Besmer P, Antonescu CR. Multiple gastrointestinal stromal tumors in type I neurofibromatosis: a pathologic and molecular study. Mod Pathol 2005; 18: 475-84. [ Links ]
60. Kinoshita K, Hirota S, Isozaki K, Ohashi A, Nishida T, Kitamura Y, et al. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol 2004; 202: 80-5. [ Links ]
61. Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 2006; 15: 1015-23. [ Links ]
62. Yamamoto H, Tobo T, Nakamori M, Imamura M, Kojima A, Oda Y, et al. Neurofibromatosis type 1-related gastrointestinal stromal tumors: a special reference to loss of heterozygosity at 14q and 22q. J Cancer Res Clin Oncol 2009; 135: 791-8. [ Links ]

