










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.102 no.11 Madrid nov. 2010
Registro Andaluz de la Poliposis Adenomatosa Familiar. Análisis de los pacientes incluidos
Andalusian Registry for Familial Adenomatous Polyposis. Analysis of patients included
M. Garzón Benavides
1Unidad Clínica de Aparato Digestivo.
2Servicio de Inmunología. Hospital Universitario Virgen del Rocío. Sevilla.
3Servicio de Aparato Digestivo. Hospital Jerez de la Frontera. Cádiz.
4Unidad Clínica de Aparato Digestivo. Hospital Universitario Reina Sofía. Córdoba
Dirección para correspondencia
RESUMEN
Objetivos: Valorar las características fenotípicas y genotípicas de los pacientes incluidos en el Registro Andaluz de la poliposis adenomatosa familiar, la relación genotipo/fenotipo y el impacto del Registro en la frecuencia de cáncer colorrectal de los familiares registrados.
Material y métodos: Estudio descriptivo de 77 pacientes con PAF, pertenecientes a 33 familias, incluidos en una base de datos centralizada a la que tienen acceso los responsables de los hospitales participantes, previa firma de cartas de confidencialidad. Todos los estudios genéticos se realizan en el Servicio de Inmunología de nuestro Hospital.
Resultados: 77 pacientes registrados (50,6% varones): 31 probandos, edad media: 32 años (13-51) y 46 familiares afectos, edad media 21,8 años (6-55). Estudio genético informado en 68/77 con resultado positivo en 92,6%. Cáncer colorrectal al diagnóstico en diez probandos (32,2%) y 2 familiares afectos (4,3%), diferencia estadísticamente significativa (p < 0,05). Se observó afectación de tramos altos en 30/61 (49%), tumor desmoides en 7/77 (9,1%) e hipertrofia del epitelio pigmentario de la retina en 23/35 (65,7%). El 86,7% de los pacientes con esta alteración mostraron mutaciones entre los codones 454 y 1.019, relación estadísticamente significativa (p < 0,05).
Conclusiones: El Registro Andaluz ha permitido ofrecer el diagnóstico genético en todas las familias afectas independientemente de su provincia de origen, mejorar el cribado, iniciar medidas preventivas precozmente y disminuir la frecuencia de cáncer colorrectal al diagnóstico en los familiares afectos registrados. La relación de la hipertrofia congénita del epitelio pigmentario de la retina con determinadas mutaciones es la única relación feno-genotípica con significación estadística.
Palabras clave: Poliposis adenomatosa familiar. Registro de PAF. Gen APC. Consejo genético. Estudio genético. Cáncer hereditario.
ABSTRACT
Objective: To evaluate the phenotype and genotype characteristic of patients included in the Andalusian Registry for familial adenomatous polyposis, the genotype/phenotype correlation and the impact of Registry in the frequency of colorectal cancer of registered.
Material and methods: A descriptive study of 77 patients with FAP belonging to 33 families, included in a centralized database visited by the physicians of the hospitals taking part in the present study, on prior signing of confidentiality letters. All genetic studies were carried out in the Immunology Service of our institution.
Results: We have included in our study 77 patients of 33 families; 31 probands with a mean age of 32 years (13-51) and 46 relatives at risk with a mean age of 21.8 years (6-55). Genetic study informed in 68/77 with positive result in 92.6%. Ten probands showed colorectal cancer (CRC) at the time of diagnosis (32.2%). Only two affected relatives showed CRC at diagnosis (4.3%), a statistically significant difference (p < 0.05). Gastrointestinal involvement was observed in 30/61 (49%), desmoid tumors in 7/77 (9.1%) and congenital hypertrophy of the retinal pigment epithelium in 23/55 (65.7%). 86.7% of patients with this alteration showed mutations between codons 454 and 1019, with a statistically significant correlation ((p< 0.05).
Conclusions: The registry has facilitated the genetic diagnosis for all affected families disregard their province of origin. It has also improved the screening of affected relatives and has made it possible to take preventive measures immediately, therefore diminishing the incidence of CRC at diagnosis in registered affected relatives. The correlation between congenital hypertrophy of the retinal pigment epithelium with some mutations is the only phenotypic-genotypic correlation with statistical significance.
Key words: Familial adenomatous polyposis. Phenotype. Genotype. FAP registry. APC gene. Genetic counseling. Genetic study. Hereditary cancer.
Introducción
La poliposis adenomatosa familiar (PAF) es una enfermedad hereditaria, autosómica dominante debida a una mutación en la línea germinal del gen APC (gen de la poliposis coli) (1). Se caracteriza por el desarrollo de múltiples pólipos en el colon y recto, generalmente más de 100, que malignizan en el 100% de las personas afectas a edades tempranas (35-40 años) (1). Una variante es la poliposis adenomatosa familiar atenuada (PAFA), caracterizada por una edad de aparición más tardía (unos 10 años) con menor número de pólipos (10-100) de predominio en colon derecho (2,3).
Existen manifestaciones extra colónicas, como la afectación gastroduodenal que aparece en un 90-100% de los pacientes, con predominio en duodeno (4,5). Entre un 4-12% de estos pacientes van a sufrir malignización, sobre todo en el área periampular (6). Especial mención merece el tumor desmoides (TD) por ser la neoplasia extra-colónica más frecuente en estos pacientes (8-12%) y segunda causa de muerte en los pacientes con colectomía profiláctica (7). Otra manifestación importante es la hipertrofia congénita del epitelio pigmentado de la retina (HCEPR), aparece en un alto porcentaje de enfermos con PAF (hasta el 80% en algunas series) y cuando son múltiples y bilaterales tienen capacidad sensitiva de screening para la misma (3).
Se trata por tanto de una enfermedad compleja, que obliga a actuar en el entorno familiar por su carácter hereditario e iniciar medidas de prevención y tratamiento precozmente. La creación de Registros de familias con PAF en otros países y comunidades ha permitido disminuir la frecuencia de CCR y mejorar la supervivencia de estos pacientes (1). Consideramos necesario ofrecer estos resultados a toda la población andaluza.
Se crea así en 2006 el Registro Andaluz de la PAF (RAPAF) con los objetivos de ofrecer el estudio genético a todas las familias afectas de nuestra comunidad, facilitar y unificar el diagnóstico y tratamiento de todos los individuos afectos.
Tras más de tres años de funcionamiento, nuestro objetivo en este estudio es conocer las características fenotípicas y genotípicas de la PAF en Andalucía, la relación genotipo-fenotipo y el impacto de la creación del registro en la reducción de la frecuencia de CCR en los familiares afectos diagnosticados por cribado.
Material y métodos
Se ha realizado un estudio descriptivo de los pacientes con PAF incluidos en el RAPAF. La creación del Registro se planteó en 2003 entre la consulta de prevención de CCR del Servicio de Aparato Digestivo del Hospital Virgen del Rocío de Sevilla y el Servicio de Inmunología del mismo hospital. Se contactó telefónicamente con los Servicios de Digestivo de 14 Hospitales de las 8 provincias andaluzas para informarles del proyecto. El Registro fue publicitado en reuniones de la Sociedad Andaluza del Patología Digestiva (SAPD) donde se nombraron los médicos responsables de los distintos Hospitales participantes.
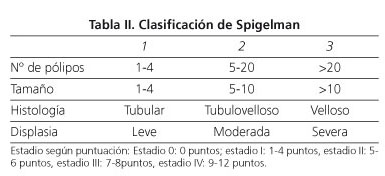
Con el apoyo del Servicio de Documentación Clínica de nuestro Hospital se creó la página Web que contiene la base de datos centralizada. En enero de 2006 comenzaron a incluirse de forma protocolizada los primeros pacientes. Para su inclusión, los pacientes debían presentar criterios clínicos y/o moleculares de PAF. Las variables recogidas fueron: edad, sexo, antecedentes familiares de PAF, tipo de intervención quirúrgica profiláctica (colectomía total con anastomosis ileorectal, colectomía subtotal con anastomosis ileorectal, ileostomía definitiva) presencia de CCR al diagnóstico, estadiaje del CCR según la clasificación TNM, tratamiento realizado, afectación de tramos altos, su estadiaje según la clasificación de Spigelman, tratamiento realizado (polipectomía, ampulectomía, pancreatectomía cefálica con preservación de píloro, etc.), manifestaciones extraintestinales (TD, HCEPR, tumor de tiroides, hepatoblastoma), desarrollo de cáncer de muñón rectal, su estadiaje según TNM, tratamiento quirúrgico realizado y características de las mutaciones encontradas.
El acceso a la base de datos se realiza a través de una clave de acceso única para cada médico, adquirida previa firma de una carta de confidencialidad, aplicando así los principios de la Ley Orgánica de Protección de Datos de Carácter Personal (LOPDP).
Para asegurar el buen funcionamiento del Registro y uso adecuado de sus datos se creó el Comité Científico, constituido por los médicos responsables de los Hospitales con más participación (Hospital Virgen del Rocío de Sevilla, Hospital Reina Sofía de Córdoba y Hospital de Jerez de la Frontera de Cádiz, y el Servicio de Inmunología del Hospital Virgen del Rocío).
Hemos realizado análisis descriptivo de las variables cualitativas y cuantitativas. El análisis comparativo de las variables cualitativas se realizó mediante el test Chi-cuadrado, considerando significación estadística cuando p < 0,05. Se ha empleado el programa estadístico SPSS 13.0 (SPSS Inc. Headquarters, Chicago, Illinois, USA).
Resultados
Desde enero de 2006 hasta junio de 2009, han participado en el RAPAF 4 de los 14 hospitales inicialmente invitados (Hospital Reina Sofía de Córdoba, Hospital de Jerez de la Frontera de Cádiz, Hospital de Torrecárdenas de Almería, Hospital Virgen del Rocío de Sevilla), lo que supone una participación del 28,5%.
En este período se han incluido 77 pacientes pertenecientes a 33 familias con una edad media de 26 años (rango 13-51 años) (50,6% varones, 49,4% mujeres). Treinta y uno (40,3%) eran probandos (edad media de 32 años; rango: 13-53 años) y 46 (59,7%) familiares afectos diagnosticados por cribado (edad media de 22 años; rango: 6-55 años).
El porcentaje de CCR en el momento del diagnóstico, ha sido del 32,3% en los probandos (10/31), y del 4,3% en los familiares afectos (2/46), diferencia estadísticamente significativa (p < 0,05). Los dos familiares afectos, estaban asintomáticos al diagnóstico y tenían edades más avanzadas que el resto de familiares (32 y 42 años). El CCR en el 40% de los probandos (4/10) estaba en un estadio TNM avanzado (IV), y en los 2 familiares en riesgo el estadio era intermedio (I y IIIB). Sólo un probando desarrolló cáncer de muñón rectal tras abandono del seguimiento durante dos años. No ha habido casos de cáncer de muñón rectal entre los familiares afectos (Tabla I).

En 61 pacientes se investigó la afectación de estómago y duodeno. Se encontraron pólipos en el 49,1% (58,3% en probandos vs. 43,2% en familiares afectos). El duodeno fue la localización más frecuente de los pólipos, 66,6% de pacientes, siendo las cifras algo más altas en probandos que en familiares afectos (71,4 vs. 62,5%). Le siguió el área peripapilar con un 50% de pacientes (probandos 64,3 vs. 37,5% familiares afectos). Los pólipos se localizaron en fundus en el 43,3%, con mayor frecuencia en los familiares afectos (50 vs. 35,7% en probandos). Un probando desarrolló cáncer duodenal a los 10 años de haber abandonado los controles de seguimiento. La gravedad de la afectación duodenal se catalogó teniendo en cuenta el estadiaje de Spigelman (Tabla II). No se obtuvo diferencia estadísticamente significativa para la localización ni la severidad de la afectación de tramos altos entre ambos grupos de pacientes (Tabla I).
Treinta y cinco de los 77 pacientes tenían estudio de fondo de ojo, y 23 de ellos (65,7%) mostraban HCEPR (Tabla III).

Siete de los 77 pacientes (9,1%), desarrollaron TD. Se localizaron en el mesenterio en 4 y en la pared abdominal, sobre cicatrices de laparotomía en 3 (Tabla III).
Se ha realizado estudio genético a 74 de los 77 pacientes. De los 68 pacientes con el estudio finalizado se ha encontrado la mutación en 63 (92,6%). El 61% de las mutaciones se localizan en el exon 15. Se han encontrado 11 mutaciones nuevas previamente no descritas. En la tabla IV se recogen los tipos de mutaciones encontradas.
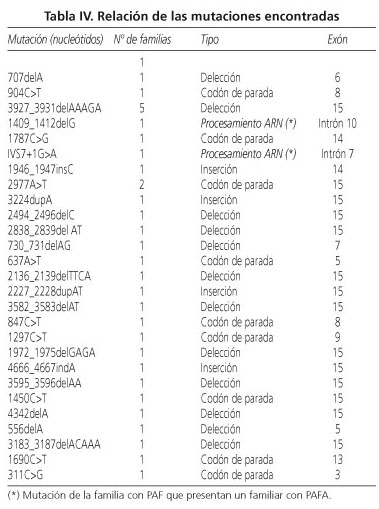
El estudio estadístico de las posibles correlaciones entre los tipos de mutaciones encontradas y las distintas manifestaciones de la PAF (colon, extracolónicas, y extradigestivas: TD y HCEPR) resultó estadísticamente significativa para la HCEPR ya que el 86,7% de los pacientes con HCEPR presentaban la mutación entre los codones 454 y 1019, relación estadísticamente significativa (p < 0,05). El resto de correlaciones entre fenotipo y genotipo no fueron significativas.
Entre los pacientes registrados sólo hay dos casos de poliposis adenomatosa familiar atenuada (PAFA), ambos en el seno de familias con PAF clásica (Tabla IV).
Discusión
Las características de la PAF, su carácter grave e incurable a pesar de tratamiento preventivo precoz, el seguimiento endoscópico estricto que precisan los pacientes, y su carácter hereditario, determina que sea fundamental el consejo y estudio genético.
La importancia del estudio genético consiste en permitir la exclusión de la enfermedad en los familiares no portadores y por tanto el riesgo de trasmitirla a su descendencia, con el consiguiente beneficio psicológico, sanitario y social. Se trata además de una medida coste-eficaz como demuestran algunos trabajos (8).
Dado que no todos los profesionales sanitarios proceden de forma adecuada en el consejo genético y saben interpretar los resultados de un estudio genético (9,10) es imprescindible crear consultas especializadas que centralicen a este tipo de pacientes (11). Por la baja incidencia de la enfermedad, algunas consultas manejan pocos pacientes, siendo necesaria la creación de grupos de colaboración a nivel regional, nacional e incluso internacional, que permitan unificar medidas de actuación (11) y realizar análisis y consejo genético adecuado (10).
Surgen así los primeros registros, el primero conocido es el del Hospital de San Marcos de Londres (12,13) cuyos objetivos eran: realizar el seguimiento a todos los familiares en riesgo, asegurar su estudio en el momento adecuado y aumentar el conocimiento de esta enfermedad (14).
Lockhart-Mummery fue el primero en indicar que el registro y seguimiento de las familias con poliposis tenía efecto beneficioso en la reducción del número de pacientes con CCR (15). Posteriormente han sido múltiples los Registros que afirman que la centralización de las familias en registros y la coordinación en el seguimiento y tratamiento de las mismas, permite disminuir la incidencia de CCR y mejorar la supervivencia (1,16,17).
Desde la creación del RAPAF se ha realizado consejo genético y ofrecido estudio genético a todos los familiares en riesgo registrados, independientemente de la provincia de origen.
Se ha encontrado la mutación en el 92,6% de los pacientes estudiados, resultados superiores al de otras series, en las que varía entre el 30-85% (18). Al valorar la relación genotipo-fenotipo sólo hemos obtenido relación entre la localización de la mutación y la presencia de HCEPR. Otros estudios han obtenido también relación entre determinadas mutaciones y el número de pólipos en la pieza de colectomía, número y gravedad de los adenomas duodenales y gástricos y la presencia de TD (19,20). De esta forma, en algunos trabajos se afirma que la localización de la mutación puede determinar el tipo de intervención quirúrgica e incluso retrasar la misma en pacientes con riesgo esperado para este tumor (18). Sin embargo la gran variabilidad fenotípica dentro de la misma familia sugiere que debe existir influencia de genes modificadores o incluso factores ambientales responsables de las características fenotípicas de los distintos miembros de la familia (18,21). La opinión mas extendida es que si bien la genética es fundamental para el diagnóstico molecular, tiene limitado valor para predecir el curso de la enfermedad y la decisión quirúrgica (18).
De los resultados del registro se extrae que la invitación a estudio de todos los familiares en riesgo ha permitido identificar a familiares afectos a edades más tempranas (22 años de los familiares afectos vs. 32 años de los probandos), iniciar las medidas preventivas a edades más precoces y disminuir la frecuencia de CCR entre los familiares afectos, 2 familiares afectos (4,3%) frente a los 10 probandos (32%), diferencia estadísticamente significativa (p < 0,05). Se observa también diferencia en el estadiaje de los tumores, más avanzado en los probandos, en los que el 40% tenían un estadio IV al diagnóstico frente a los estadios inicial e intermedio (I y IIIB respectivamente) de los familiares afectos.
La afectación de tramos altos en los pacientes registrados ha sido de 49% con predominio de la afectación en duodeno (66,6%), al igual que se recoge en otros trabajos (4,6,22-25).
En nuestra serie, la manifestación extraintestinal más frecuente, la HCEPR se encontró en el 65,7% de los pacientes estudiados, porcentaje más bajo que el de otras series donde llega a alcanzar el 80% (26).
El TD se ha observado en el 9,1% de nuestros pacientes, frecuencia similar a otros trabajos (27). Es importante diagnosticarlo y tratarlo precozmente por ser la segunda causa de muerte en pacientes con colectomía profiláctica (7,28). Se han mencionado determinadas mutaciones, como posibles factores de riesgo para desarrollarlo. Ninguno de nuestros pacientes con TD tenían la mutación en los lugares descritos como asociados al tumor (27,29) por lo que consideramos que lo fundamental para su diagnóstico precoz es la realización de palpación abdominal y resonancia magnética abdominal, al ser la pared abdominal y el mesenterio las localizaciones más frecuentes (27).
Dos de las familias registradas presentan un familiar afecto con PAFA. Esta diferencia de fenotipos dentro de una misma familia a pesar de la misma mutación, podría ser debido a que las mutaciones encontradas afectan al procesamiento del ARN mensajero. Este tipo de mutaciones implica una disminución en la eficiencia de procesamiento del ARN mensajero (30) y esta mayor o menor eficiencia explicaría los distintos fenotipos en miembros de la misma familia.
Finalmente la aparición de cáncer intestinal durante la evolución de la enfermedad solo ha estado asociada en nuestros casos con el abandono del seguimiento y no con el tipo de mutación de estos pacientes.
Algunos de nuestros resultados tales como la imposibilidad para correlacionar el TD, la gravedad de la afectación de tramos altos y el desarrollo de cáncer de muñón rectal con determinadas mutaciones descritas en la literatura pueden verse afectados por el bajo tamaño muestral debido en parte a la baja participación de los hospitales invitados. La creación de consultas especializadas en prevención de CCR y la comprensión de la necesidad de registrar a todos las familias afectas quizá nos ayude en los próximos años a obtener todos y no sólo algunos de los objetivos propuestos.
 Dirección para correspondencia:
Dirección para correspondencia:
Marta Garzón Benavides.
Servicio de Aparato Digestivo.
Hospital Universitario Virgen del Rocío.
Avda. Manuel Siurot, s/n.
41013 Sevilla.
e-mail: mgarzonb@hotmail.com
Recibido: 18-03-10.
Aceptado: 07-07-10.
Bibliografía
1. Morton DG, Macdonald F, Haydon J, Cullen R, Barker G, Hulten M, et al. Screening practice for familial adenomatous polyposis: the potential for regional registers. Br J Surg 1993; 80: 255-8. [ Links ]
2. Lynch HT, Smyrk T, McGinn T, Lanspa S, Cavalieri J, Lynch J, et al. Attenuated familial adenomatous polyposis (AFAP). A phenotypically and genotypically distinctive variant of FAP. Cancer 1995; 76: 2427-33. [ Links ]
3. Elizabeth Half, Dani Bercovich and Paul Rozen. Familial adenomatous polyposis. Orphaned J Rare Dis 2009; 4: 22. [ Links ]
4. Wallace MH, Phillips RK. Upper gastrointestinal disease in patients with familial adenomatous polyposis. Br J Srg 1998; 85: 742-50 [ Links ]
5. Brosens LA, Keller JJ, Offerhaus GJ, Goggins M, Giardiello FM. Prevention and management of duodenals polyps in Familial Adenomatous Polyposis. Gut 2005; 54: 1034-43. [ Links ]
6. Morpurgo E, Vitale C, Galandiuk S, Kimberling J, Ziegler C, Polk HC Jr. Clinical characteristics of Familial Adenomatous Polyposis and Management of duodenal adenomas. J. Gastointest Surg 2004; 8(5): 559-64 [ Links ]
7. Griffioen G, Bus PJ, Vasen HF, Verspaget HW, Lamers CB. Extracolonic manifestations of familial adenomatous polyposis: desmoids tumors, and upper gastrointestinal adenomas and carcinomas. Scand J Gastroenterol Suppl 1998; 225: 85-91. [ Links ]
8. Olry De Labry Lima A, Sordo del Castillo L, García Mochon L, Epstein D, Bermudez Tamayo C, Villegas Portero R. An economic assessment of genetic testing for familial adenomatous polyposis. Rev Esp Enferm Dig 2008; 100(8): 470-5. [ Links ]
9. Giardello FM, Bresinger JD, Peterson GM, Luce MC, Hylind LM, Bacon JA, et al. The use and interpretation of comercial APC gene testing for familial adenomatous polyposis. N Engl J Med 1997; 336: 823-7. [ Links ]
10. Fernández-Suárez A, Cordero-Fernández C, García Lozano R, Pizarro A, Garzón M, Núñez Roldán A. Implicaciones clínicas y éticas del consejo genético en la poliposis adenomatosa familiar. Rev Esp Enferm Dig 2005; 97(9): 654-65. [ Links ]
11. Church J, Kiringoda R, LaGuardia L. Inherited colorectal cancer Registries in The United States: the State of the Union. Dis Colon Rectum 2004; 47: 674-8. [ Links ]
12. Obrador A, Thomson JPS. El Registro de Poliposis del Hospital de San Marcos de Londres. Gastroenterol Hepatol 2002; 25(4): 267-71. [ Links ]
13. Spiegelman AD, Thomson JP. Introduction, history and registries in familial adenomatous polyposis. London, Edward Arnold, 2004. [ Links ]
14. Bussey HJR. St Mark's Hospital Polyposis Register. Annual Report St Mark's Hospital 1970: 20-1. [ Links ]
15. Lockhart-Mummery P. Cancer and Heredity. Lancet 1925; 1: 427-9. [ Links ]
16. Reyes Moreno J, Ginard Vicens D, Vanrell M, Mariño Z, Garau I, Llompart A, Obrador Adrover A. Mejoría de la supervivencia de la poliposis adenomatosa familiar después del establecimiento de un registro. Med Clin (Barc). 2007; 129(2): 51-2. [ Links ]
17. Bülow S, Vulgo C, Faurschou Nielsen T, Karlsen & Moesgaard L. Centralized Registration, Prophylactic examination and treatment results in improved prognosis in familial adenomatous polyposis. Scand J Gastroenterol 1995; 30: 989-93. [ Links ]
18. Friedl W, Caspari R, Sengteller M, Uhlhaas S, Lamberti C, Jungck M, et al. Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut 2001; 48: 515-21. [ Links ]
19. Caspari R, Olschwang S, Friedl W, Mandl M, Boisson C, Böker T, et al. Familial adenomatous polyposis: desmoids tumors and lack of ophthalmic lesions (CHRPE) associated with APC mutation beyond codon 1444. Hum Mol Genet 1995; 4: 337-40. [ Links ]
20. Enomoto M, Konishi M, Iwama T, Utsunomiya J, Sugihara K, Miyaki M. The relationship between frequencies of extracolonic manifestations and the position of APC germline mutation in patients with familial adenomatous polyposis. JPN J Clin Oncol 2000; 30(2): 82-8. [ Links ]
21. Crabtree MD, Tomlison IMP, Hodgson SV, Neale K, Phillips RKS, Houlston RS. Explaining variation in familial adenomatous polyposis: relationship between genotype and phenotype and evidence for modifier genes. Gut 2002; 51: 420-3. [ Links ]
22. Kashiwagi H, Spigelman AD. Gastroduodenal lesions in familial adenomatous polyposis. Surg Today 2000; 30: 675-82. [ Links ]
23. Jalving M, Koornstra J, Götz J, Van der Waaij LA, de Jong S, Zwart N, et al. High-grade dysplasia in sporadic fundic gland polyps: a case report and review of the literature. Eur Journal of Gastroent and Hepatol 2003; 15: 1229-33. [ Links ]
24. Hofgärtner W, Thorp M, Ramus M, Delorefice G, Chey WY, Ryan CK, et al. Gastric adenocarcinoma associated with fundic gland polyps in patient with attenuated familial adenomatous polyposis. Am Journal of Gastroenterology 1999; 94: 2275-81. [ Links ]
25. Cordero-Fernández C, Garzón-Benavides M, Pizarro-Moreno A, García-Lozano R, Márquez-Galán JL, López-Ruiz T, et al. Gastroduodenal involvement in patients with familial adenomatous polyposis. Prospective study of the nature and evolution of polyps: evaluation of the treatment and surveillance methods applied. Eur J Gastroenterol Hepatol 2009; 21 (10): 1161-7. [ Links ]
26. Vasen HFA, Möslein G, Alonso A, Aretz S, Berstein I, Bertario L, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57: 704-13. [ Links ]
27. Pikaar A, Notier JW, Griffioen G, Vasen HF. Desmoids tumors in patients with familial adenomatous polyposis. Ned Tijdschr Geneeskd 2002; 146: 1355-9. [ Links ]
28. Arvanitis ML, Jagelman DG, Fazio VW, Lavery IC, McGannon E. . Mortality in patients with familial adenomatous polyposis. Dis Colon Rectum 1990; 33: 639-42. [ Links ]
29. Nieuwenhuis MH, Vasen HFA. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol 2007; 61: 153-61. [ Links ]
30. Aretz S, Uhlhaas S, Sun Y, Pagenstecher C, Mangold E, Caspari R, Möslein G, et al. Familial adenomatous polyposis: aberrant splicing due to missense or silent mutations in the APC gen. Hum Mutat 2004; 24(5): 370-80. [ Links ]

