










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versão impressa ISSN 1130-0108
Rev. esp. enferm. dig. vol.103 no.7 Madrid Jul. 2011
https://dx.doi.org/10.4321/S1130-01082011000700010
Mutaciones en los genes HFE y TFR2 en un paciente español con hemocromatosis
Mutations in HFE and TFR2 genes in a Spanish patient with hemochromatosis
Alejandro del Castillo Rueda1, Nuria Cuadrado Grande2, Emilio Álvarez Fernández3, Rafael Enríquez de Salamanca1, Luis Antonio Álvarez Sala1 y María Josefa Morán Jiménez2
1Unidad de Ferropatología. Departamento de Medicina Interna. Hospital General Universitario Gregorio Marañón. Madrid.
2Centro de Investigación. Instituto de Investigación. Hospital 12 de Octubre. Madrid.
3Departamento de Anatomía Patológica. Hospital General Universitario Gregorio Marañón. Madrid
Financiación obtenida del "Fondo de Investigaciones Sanitarias" (FIS 07/0074) y la "Fundación Mutua Madrileña de Investigación Biomédica" (FMM 2007-73). Los autores agradecen el soporte técnico del Servicio de Genómica del "Instituto de Investigación del Hospital 12 de Octubre".
Dirección para correspondencia
RESUMEN
La enfermedad por sobrecarga de hierro está originada por diversas anomalías genéticas. El estudio genético de esta enfermedad confirma su carácter hereditario y nos permite ofrecer consejo genético a los familiares en primer grado. Hemos realizado resonancia magnética y biopsia de hígado en un paciente asintomático con más de 1.000 µg/l de ferritina en suero, y hemos analizado los genes implicados en el metabolismo del hierro. El fenotipo de sobrecarga de hierro se confirmó por la presencia de un patrón de depósito de hierro en el hígado con predominio periportal que sugiere la existencia de una enfermedad genética. En el caso que presentamos, el estudio genético reveló que el paciente es doble heterocigoto para las mutaciones c.187C>G (p.H63D) y c.840C>G (p.F280L) en los genes HFE y receptor 2 de transferrina (TFR2), respectivamente.
Palabras clave: Hemocromatosis. HFE. Receptor 2 de la transferrina.
ABSTRACT
Iron overload disease has a wide variety of genotypes. The genetic study of this disease confirms its hereditary nature and enables us to provide genetic counseling for first-degree relatives. We performed magnetic resonance imaging and liver biopsy in an asymptomatic patient with more than 1,000 µg/L of serum ferritin and studied the genes involved in this condition. The phenotype of iron overload is confirmed by a predominantly periportal pattern of iron deposits in the liver suggestive of genetic disease. In the case we present the molecular study revealed a double heterozygosity for the mutations c.187C>G (p.H63D) and c.840C>G (p.F280L) in the HFE and transferrin receptor 2 (TFR2) genes, respectively.
Key words: Hemochromatosis. HFE. Transferrin receptor 2.
Introducción
El síndrome clínico de la hemocromatosis se conoce desde hace más de 100 años. Sin embargo, los avances más importantes en el conocimiento de esta enfermedad se han producido en las últimas dos décadas, especialmente con el descubrimiento del gen HFE (1). La hemocromatosis presenta una marcada heterogeneidad en cuanto a la etiología genética, tipo de herencia, factores ambientales, y manifestaciones clínicas y analíticas (2-5).
Aunque la mutación c.845G>A (p.C282Y) en homocigosis en el gen HFE es responsable de la forma clásica de hemocromatosis hereditaria, conocida como hemocromatosis tipo 1, otros genotipos, tales como la heterocigosis compuesta c.845G>A (p.C282Y)/c.187C>G (p.H63D) en el gen HFE, están asociados con la sobrecarga de hierro (6). La mutación c.187C>G (p.H63D) en heterocigosis no parece ser responsable de la sobrecarga de hierro o hemocromatosis; por lo tanto, si aparecen datos clínicos y bioquímicos de hemocromatosis con este genotipo, es necesario analizar factores asociados como la ingesta de alcohol, virus de la hepatitis o mutaciones en otros genes implicados en la homeostasis del hierro (7,8). En algunos casos, la aparición simultánea de alteraciones en los genes HFE y TFR2 en pacientes con sobrecarga de hierro son responsables de la hemocromatosis heredada digénicamente (9,10).
La hemocromatosis tipo 3 está causada por mutaciones en el gen del receptor 2 de transferrina (TFR2). Los primeros casos se describieron en dos familias sicilianas con la mutación c.750C>G (p.Y250X) en homocigosis en este gen (11). El receptor 2 de transferrina es homólogo al TFR1, se expresa principalmente en el hígado y la afinidad de la proteína TFR2 por la transferrina es 30 veces superior que la de TFR1 (11). Los pacientes con mutaciones en heterocigosis en el gen TFR2 no tienen sobrecarga de hierro, a diferencia de los pacientes con mutaciones en heterocigosis en el gen HFE. Las manifestaciones clínicas de la hemocromatosis tipo 3 son comparables a las de tipo 1, aunque hay informes de casos más severos con una edad de aparición más temprana (12).
Presentamos el caso de un paciente con mutaciones en los genes HFE y TFR2 y un fenotipo clínico de sobrecarga de hierro. Este caso confirma la heterogeneidad genotípica y fenotípica de la hemocromatosis.
Caso clínico
Un hombre de 50 años de edad fue remitido a nuestra Unidad ya que desde hacía dos años tenía la concentración de ferritina en suero superior a 1.000 µg/l (rango normal, 26-370). El paciente tenía la ferritina sérica (1.277 µg/l) y la γGT (116 U/L; rango normal, 10-60) aumentadas, y la ecografía del hígado nos llevó a sospechar posible hígado graso. Las pruebas analíticas y serológicas complementarias no revelaron ninguna anomalía. El paciente estaba asintomático. No bebía alcohol ni consumía tabaco. Su historia clínica personal y familiar, y el examen físico no reflejaban nada destacable.
Analizamos los marcadores del metabolismo del hierro (niveles de hierro en suero, ferritina, receptor soluble de transferrina e índice de saturación de transferrina), y la concentración de hierro hepático y la arquitectura del tejido hepático mediante una biopsia del hígado. Las secciones de tejido hepático de cinco micrómetros se tiñeron con hematoxilina-eosina, y azul Prusia de Perls para detectar la presencia de hierro. Se estudió la imagen obtenida mediante resonancia magnética del hígado (MRI). También se buscaron las variaciones alélicas c.845G>A (p.C282Y) y c.187C>G (p.H63D) en el gen HFE.
El ADN genómico se extrajo de una muestra de sangre periférica, la reacción en cadena de polimerasa (PCR) se realizó mediante oligonucleótidos específicos para amplificar las regiones codificantes, las regiones intrónicas adyacentes, y las regiones sin traducir 5 ' y 3 ' (UTR) de los genes: HFE, hemojuvelina (HJV), hepcidina (HAMP), receptor 2 de transferrina (TFR2) y ferroportina (SLC40A1). Los productos de PCR se secuenciaron bidireccionalmente en el secuenciador de ADN ABI Prism 3130x1, y las secuencias se compararon con las de referencia (números de accesión del GenBank: NG_008720.1, NG_011568.1, NG_011563.1, NM_003227.3 y NG_009027.1, respectivamente, para los genes mencionados).
Se obtuvo el consentimiento informado del paciente según las recomendaciones locales para el estudio genético y la biopsia del hígado. El protocolo de estudio se realizó de acuerdo con los principios éticos de la declaración de Helsinki (2008).
Los parámetros de hierro del paciente fueron: ferritina en suero, 1.070 µg/l (rango normal, 26-370 µg/l); hierro en suero, 160 µg/dl (rango normal, 59-158 µg/dl); índice de saturación de transferrina, 48% (rango normal, 20-45%); y receptor soluble de transferrina, 0,90 mg/l (rango normal, 1,76 ± 0,83 mg/l). La imagen de resonancia magnética hepática mostró anomalías difusas de la intensidad en el parénquima, asociadas con un aumento de hierro; el contenido de hierro hepático fue estimado en 160 µmol/g (valores normales, < 36 µmol/g). Se realizó una biopsia hepática percutánea guiada por ecografía. La evaluación histopatológica mostró una arquitectura lobular normal, estrechos espacios portales sin inflamación o fibrosis y sin lesiones vasculares o ductales. El lóbulo estaba compuesto por hepatocitos con morfología normal y con depósitos de baja intensidad de hemosiderina, con el patrón típico de hemocromatosis, es decir, depósitos de hierro localizados casi exclusivamente en las células parenquimatosas en áreas periportales, con un patrón granular y una distribución pericanalicular (Fig. 1).
El paciente fue sometido a tratamiento con flebotomías y, después de 20 flebotomías quincenales, se eliminaron más de 5 g de hierro, y las concentraciones de γGT y ferritina volvieron a valores normales. Actualmente el paciente se somete a flebotomías de mantenimiento dos veces al año.
El estudio molecular de los genes implicados en el metabolismo del hierro mostró las siguientes variantes alélicas en estado heterocigoto: c.187C>G (p.H63D) en el gen HFE y c.840C>G (p.F280L) en el gen TFR2 (Fig. 2).
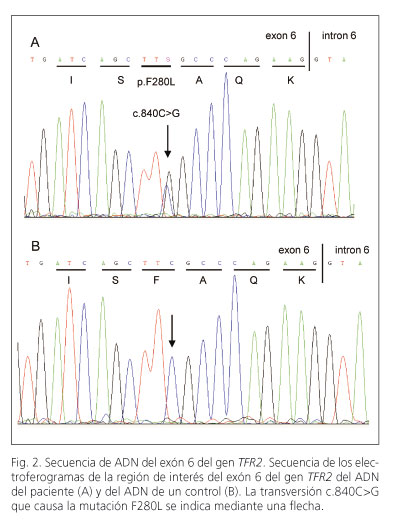
Discusión
La hemocromatosis es una enfermedad clínica y genéticamente heterogénea, causada por un aumento inadecuado de absorción intestinal de hierro. Aunque la mayoría de los pacientes son homocigotos para la mutación c.845G>A (p.C282Y) del gen HFE, algunos factores genéticos y ambientales pueden modificar el fenotipo de hemocromatosis. Los pacientes con mutaciones en doble heterocigosis c.845G>A/c.187C>G (p.C282Y/p.H63D) en el gen HFE muestran un fenotipo común de hemocromatosis y, pacientes con mutaciones en otros genes como SLC40A1, TFR2, HJV y HAMP, heredadas de forma monogénica o digénica, muestran una amplia variedad de manifestaciones clínicas (9,13).
La hemocromatosis tipo 3 es una enfermedad recesiva inusual causada por mutaciones en el gen TFR2. Esta enfermedad ha sido descrita en un pequeño grupo de familias de origen ítalo-francés, así como en familias de origen escocés, japonés y portugués, y causa hemocromatosis clínica indistinguible de la hemocromatosis tipo 1. Sin embargo, el diagnóstico de la hemocromatosis tipo 3 se realiza a una edad significativamente menor que en la hemocromatosis tipo 1 (12,14). La hemocromatosis tipo 3 es un síndrome intermedio entre la hemocromatosis tipo 1 y dos formas juveniles, hemocromatosis tipo 2a y tipo 2b, que son causadas por mutaciones en los genes HJV y HAMP; por lo tanto, las mutaciones del gen TFR2 deben tenerse en cuenta en los pacientes jóvenes con hemocromatosis (10,14,15).
La hepcidina es la hormona que controla la absorción intestinal de hierro, su baja concentración circulante es una característica común de la hemocromatosis tipo 1, 2 y 3 (16). HFE, TFR2 y HJV se consideran moduladores de la síntesis o actividad de la hepcidina, que a su vez regula la liberación de hierro en la sangre de células duodenales y macrófagos, manteniendo así los niveles de hierro apropiado para satisfacer las necesidades del organismo (17,18). La combinación de mutaciones en los genes HFE y TFR2 y mutaciones en los genes HAMP y HJV conducen a formas más graves de hemocromatosis (debido a una desregulación en la absorción intestinal de hierro), con un aumento de la entrada de hierro en la sangre y el almacenamiento de hierro en los tejidos (10).
Hay casos descritos de pacientes con hemocromatosis tipo 3 con una mutación en heterocigosis en el gen TFR2 y presencia o no de mutaciones del gen HFE. Mendes y cols. (19) describieron un paciente con hábitos alcohólicos y con dos mutaciones en heterocigosis, c.187C>G en el gen HFE y c.840C>G en el gen TFR2, las cuales eran responsables de la enfermedad de sobrecarga de hierro. Esta doble heterocigosis se observó también en el paciente que presentamos. No tenía hábitos tóxicos, y el fenotipo de sobrecarga férrica se confirmó mediante resonancia magnética y biopsia hepática. Su situación no era grave y respondió a las flebotomías. Aunque no encontramos mutaciones en otros genes estudiados en el paciente (HJV, HAMP y SLC40A1), otros factores ambientales y genéticos podrían estar involucrados en el almacenamiento de hierro en los pacientes con hemocromatosis.
La identificación temprana de los pacientes con sobrecarga de hierro es muy importante para el pronóstico. Por lo tanto, una biopsia de hígado, la medición del índice de hierro hepático y un estudio genético resultan necesarios para clarificar el diagnóstico, clasificación y tratamiento de los pacientes con hemocromatosis.
![]() Dirección para correspondencia:
Dirección para correspondencia:
María Josefa Morán Jiménez.
Centro de Investigación.
Hospital Universitario 12 de Octubre.
Avenida de Córdoba, s/n.
28041 Madrid.
e-mail: moranjimenez@h12o.es
Recibido: 21-12-10.
Aceptado: 13-01-11.
Bibliografía
1. Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996;13:399-408. [ Links ]
2. Bacon BR, Britton RS. Clinical penetrance of hereditary hemochromatosis. N Engl J Med 2008;358:291-2. [ Links ]
3. Barton JC, Lafreniere SA, Leiendecker-Foster C, Li H, Acton RT, Press RD, et al. HFE, SLC40A1, HAMP, HJV, TFR2, and FTL mutations detected by denaturing high-performance liquid chromatography after iron phenotyping and HFE C282Y and H63D genotyping in 785 HEIRS Study participants. Am J Hematol 2009;84:710-4. [ Links ]
4. Jorquera F, Dominguez A, Diaz-Golpe V, Espinel J, Muñoz F, Herrera A, et al. C282Y and H63D mutations of the haemochromatosis gene in patients with iron overload. Rev Esp Enferm Dig 2001;93:293-302. [ Links ]
5. Solis-Herruzo JA, Solis Muñoz P. Non-HFE hemochromatosis. Rev Esp Enferm Dig 2005;97:266-86. [ Links ]
6. Pietrangelo A. Inherited metabolic disease of the liver. Curr Opin Gastroenterol 2009;25:209-14. [ Links ]
7. Rochette J, Le Gac G, Lassoued K, Ferec C, Robson KJ. Factors influencing disease phenotype and penetrance in HFE haemochromatosis. Hum Genet 2010;128:233-48. [ Links ]
8. Ropero Gradilla P, Villegas Martinez A, Fernández Arquero M, García-Agúndez JA, González Fernández FA, Benítez Rodríguez, et al. C282Y and H63D mutations of HFE gene in patients with advanced alcoholic liver disease. Rev Esp Enferm Dig 2001;93:156-63. [ Links ]
9. Merryweather-Clarke AT, Cadet E, Bomford A, Capron D, Viprakasit V, Miller A, et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum Mol Genet 2003;12:2241-7. [ Links ]
10. Pietrangelo A, Caleffi A, Henrion J, Ferrara F, Corradini E, Kulaksiz H, et al. Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes. Gastroenterology 2005;128: 470-9. [ Links ]
11. Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet 2000;25:14-5. [ Links ]
12. Aguilar-Martinez P. Non-HFE-related hereditary iron overload. Presse Med 2007;36:1279-91. [ Links ]
13. Brissot P, Troadec MB, Bardou-Jacquet E, Le Lan C, Jouanolle AM, Deugnier Y, et al. Current approach to hemochromatosis. Blood Rev 2008;22:195-210. [ Links ]
14. Biasiotto G, Camaschella C, Forni GL, Polotti A, Zecchina G, Arosio P. New TFR2 mutations in young Italian patients with hemochromatosis. Haematologica 2008;93:309-10. [ Links ]
15. Gerolami V, Le Gac G, Mercier L, Nezri M, Berge-Lefranc JL, Ferec C. Early-onset haemochromatosis caused by a novel combination of TFR2 mutations (p.R396X/c.1538-2 A>G) in a woman of Italian descent. Haematologica 2008;93:e45-6. [ Links ]
16. Pelucchi S, Mariani R, Trombini P, Coletti S, Pozzi M, Paolini V, et al. Expression of hepcidin and other iron-related genes in type 3 hemochromatosis due to a novel mutation in transferrin receptor-2. Haematologica 2009;94:276-9. [ Links ]
17. De Domenico I, Ward DM, Kaplan J. Hepcidin regulation: ironing out the details. J Clin Invest 2007;117:1755-8. [ Links ]
18. Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell 2010;142:24-38. [ Links ]
19. Mendes AI, Ferro A, Martins R, Picanço I, Gomes S, Cerqueira R, et al. Non-classical hereditary hemochromatosis in Portugal: novel mutations identified in iron metabolism-related genes. Ann Hematol 2009;88:229-34. [ Links ]

