










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.104 no.2 Madrid feb. 2012
https://dx.doi.org/10.4321/S1130-01082012000200007
PICTURES IN DIGESTIVE PATHOLOGY
External and internal appearance of hepatorenal polycystic disease
Apariencia externa e interna de la poliquistosis hepatorrenal
Pilar Olivencia-Palomar, Susana Ávila-Nasi, Roberto González-Soler, Elena Castro and Leopoldo López Rosés
Department of Digestive Disease. Hospital Xeral-Calde. Lugo
Case report
A fifty four years old male was followed in nephrology clinic since 1999 for hepatorenal polycystic disease. The patient was referred for abdominal distension with postprandial fullness. Family history of hepatorenal polycystic disease in the father, grandmother and a paternal aunt.
On physical examination there was a globular abdomen, with multiple irregular masses, which imprint on the abdominal wall, and tenderness are hard and painless (Fig. 1). In additional studies, laboratory tests displayed normal renal and liver function.

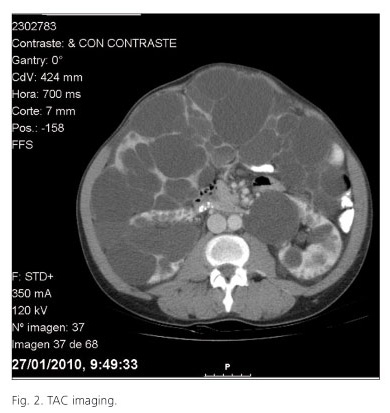
We performed a CT scan that showed an important hepatomegaly, in which virtually the entire liver parenchyma was replaced by multiple images of rounded, hypodense, cystic appearance and different sizes, in conjunction with the polycystic disease, producing a significant mass effect on gastric cavity, the duodenal and intestinal loops, which were displaced towards the pelvic region. Both kidneys were enlarged with multiple bilateral cortical cysts of various sizes (Fig 2).
Discussion
Polycystic liver disease is inherited as an autosomal dominant disease and usually occurs in combination with polycystic kidney disease or cysts in other organs including the pancreas, spleen, ovaries, and, more rarely, the lung. The most common clinical manifestations include abdominal distention with palpable mass or masses, as in this patient, early satiety, and sometimes abdominal pain, jaundice, or dyspnea. There may be compression of the portal vein from the cysts, leading to portal hypertension and its complications (ascites, esophageal variceal bleeding, etc.) (1,2). The prognosis is determined by the hepatorenal polycystic kidney disease. In case of terminal renal failure, the double transplant is a treatment to be consider (3).
References
1. Pérez Flores R, Vega García-Luengos M, Figueruela López B, Hernández Rial M, Martín Eleno M, Villaamil Cabezudo R. Hepatorenal polycystic disease in adults. Nonsurgical alternative treatment. Rev Esp Enferm Dig 1990;78(6):392-3. [ Links ]
2. Deschênes G. Hepatorenal autosomic recessive polycystic disease, a polyductine disease. Arch Pediatr 2002;9(9):1003. [ Links ]
3. Chava SP, Singh B, Stangou A, Battula N, Bowles M, O'Grady J, et al. Simultaneous combined liver and kidney transplantation. A single center experience. Clin Transplant 2010;24(3):E62-8. [ Links ]

