










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.106 no.3 Madrid mar. 2014
REVISIÓN
Regeneración hepática; el secreto mejor guardado. Una forma de respuesta al daño tisular
Liver regeneration - The best kept secret. A model of tissue injury response
Javier A.-Cienfuegos, Fernando Rotellar, Jorge Baixauli, Fernando Martínez Regueira, Fernando Pardo y José Luis Hernández Lizoáin
Departamento de Cirugía General. Clínica Universidad de Navarra. Pamplona
Dirección para correspondencia
RESUMEN
La regeneración hepática (RH) representa una de las formas más extraordinarias de respuesta al daño tisular. Debido a sus implicaciones terapéuticas, ha sido motivo de un interés extraordinario en las últimas décadas.
La RH es un proceso muy complejo y estrictamente regulado que cumple las características de los sistemas biológicos más evolucionados (robustness) y que explica la dificultad de interferir en ella con usos terapéuticos.
La RH reproduce el modelo fisiológico de respuesta al daño con una fase primera de preparación o "cebado" de los hepatocitos -transición G0-G1 del ciclo celular- y una fase ulterior de proliferación -fases S/M del ciclo- que termina con la recuperación de la masa hepática.
Dicho proceso se ha relacionado con los mediadores de la respuesta biológica al daño tisular: hormonas, sistema del complemento, plaquetas, citocinas proinflamatorias (TNF-α, IL-1β, IL-6), factores de crecimiento (HGF, EGF, VGF, etc.) y factores antiinflamatorios (Il-10, TGF-β).
Debido a su complejidad y a su estricta regulación, se explica que todavía hoy la única alternativa a la insuficiencia hepática sea el trasplante hepático.
La identificación reciente de las células pluripotenciales inducidas (iPS) y la capacidad plástica de las células madre mesenquimales (CD133+) ha suscitado nuevas expectativas a la terapia celular regenerativa. Dichos trabajos han confirmado la cooperación entre las células mesenquimales y epiteliales.
En el presente trabajo se describen los mecanismos fisiológicos de la regeneración hepática.
Palabras clave: Regeneración hepática. Respuesta inflamatoria estéril. Ciclo celular. p53. Respuesta estrés hiperproliferativo. Células pluripotenciales inducidas (iPS).
ABSTRACT
Liver regeneration (LR) is one of the most amazing tissue injury response. Given its therapeutic significance has been deeply studied in the last decades.
LR is an extraordinary complex process, strictly regulated, which accomplishes the characteristics of the most evolutionary biologic systems (robustness) and explains the difficulties of reshaping it with therapeutic goals.
TH reproduces the physiological tissue damage response pattern, with a first phase of priming of the hepatocytes -cell-cycle transition G0-G1-, and a second phase of proliferation -cell-cycle S/M phases- which ends with the liver mass recovering.
This process has been related with the tissue injury response regulators as: complement system, platelets, inflammatory cytokines (TNF-α, IL-1β, IL-6), growth factors (HGF, EGF, VGF) and anti-inflammatory factors (IL-10, TGF-β).
Given its complexity and strict regulation, illustrates the unique alternative to liver failure is liver transplantation.
The recent induced pluripotential cells (iPS) description and the mesenchymal stem cell (CD133+) plastic capability have aroused new prospects in the cellular therapy field. Those works have assured the cooperation between mesenchymal and epithelial cells.
Herein, we review the physiologic mechanisms of liver regeneration.
Key words: Liver regeneration. Sterile inflammatory response. Cell-cycle. p53. Hyperproliferative stress response. Induced pluripotentianl cells (iPS).
Abreviaturas
AP-1: proteína activadora 1.
ABs: ácidos biliares.
ATP: adenosina trifosfato.
AR: amfiregulina.
ARNm: ácido ribonucleico mensajero.
Cdc25b: fosfatasa 2 inductora de fase M.
Cdk: quinasa dependiente de ciclina (del inglés: cyclin-dependent kinase).
CD117: células progenitoras hematopoyéticos.
CD133: células progenitoras endoteliales.
Ckls: inhibidores de los complejos Cdk-ciclina.
Cl4C: tetracloruro de carbono.
C-met: receptor de membrana de HGF.
CT-1: cardiotrofina 1.
C3a: fragmento proteolítico "a" de la proteína C3 del complemento.
C3b: fragmento proteolítico "b" de la proteína C3 del complemento.
C5a: fragmento proteolítico "a" de la proteína C5 del complemento (anafilotoxina).
DAMP: patrones moleculares asociados a la lesión (del inglés: damage-associated molecular pattern).
DNA: ácido desoxirribonucleico.
EGF: factor de crecimiento epidérmico.
EGFR: receptor del factor de crecimiento epidérmico.
FAH -/-: homozigosis para la mutación de la flumaril acetotato hidrolasa (tirosinemia).
Fase M: fase de mitosis.
Fase S: fase de síntesis de DNA.
FGF: factor de crecimiento fibroblástico.
FXR: receptor X farnesoide.
gp 130: glicoproteína 130.
GSK3β: quinasa de la glucógeno sintetasa 3β.
G0: fase 0: "de descanso, quiescencia del ciclo celular". Del inglés: GAP.
G1: fase 1: "preparativa del ciclo celular, entre la fase M y la fase S". Del inglés: GAP.
G2: fase 2: "preparativa del ciclo celular, entre la fase S y la mitosis". Del inglés: GAP.
HB-EGF: factor semejante al factor epidérmico ligado a la heparina.
HGF: factor de crecimiento de los hepatocitos.
HIF-1α: factor de transcripción inducido por hipoxia 1α.
HMGB1: proteína de alta movilidad del grupo de caja 1 (high movility group box 1).
Hsp: proteínas del choque térmico (heat shock proteins).
IGF-1: factor de crecimiento insulínico.
IkbKβ: inhibidor citoplasmático de NF-Kβ.
IL: interleucina.
IL-1β: interleucina 1 beta.
IL-6: interleucina 6.
IL-18: interleucina 18
IL-6R/gp130: glicoproteína 130 receptor de IL-6.
iPS: células madre pluripotenciales inducidas (induced pluripotent stem cells).
JAK: proteína tirosina quinasa Janus o Janus quinasa.
KO: animal defectivo para un gen (del inglés: knout-out).
Linfocitos NK: linfocitos citolíticos naturales (del inglés: natural killer).
LPS: lipoproteína bacteriana.
MAPK: proteína quinasa activada por mitógeno.
ME: matriz extracelular.
Myc: oncoproteína Myc.
NF-Kβ: factor nuclear Kappa β.
NKT: linfocitos citolíticos naturales.
NLRP: subfamilia de receptores tipo NOD (NLR, del inglés: NOD-like receptors: nucleotide digomerization domain - containing protein).
NOD: proteína con el dominio de oligomerización que se une a nucleótidos (del inglés: nucleotide digomerization domain - containing protein).
PAF: factor activador de las plaquetas.
PAMP: patrones moleculares asociados a microorganismos patógenos (del inglés: pathogen associated molelular patterns).
PDGF: factor de crecimiento plaquetario.
Punto R: punto de "restricción" del ciclo celular.
p53: proteína p53.
RH: regeneración hepática.
ROS: radicales libres de oxígeno.
RRP: receptores para el reconocimiento de patrones.
RTK: receptor tirosina quinasa (del inglés: receptor tyrosine kinases).
RTT: receptores tipo toll (toll-like receptors).
SOCS: supresores de proteínas transmisoras de señales de citocinas.
SOCS-3: supresor de la señalización de citocinas.
STAT3: activador de la transcripción y transductor de la señal 3 (del inglés: signal transducer and activator of transcription).
TFG-α: factor de crecimiento transformante alfa.
TNF-α: factor de necrosis tumoral alfa.
TNF-R: receptores del factor de necrosis tumoral.
TPM1: triptófano hidrolasa.
VEGF: factor de crecimiento endotelio-vascular.
"La naturaleza adapta el órgano a la función y no la función al órgano".
Artistóteles, en Sobre los Animales
Antecedentes
La regeneración hepática (RH) es uno de los fenómenos biológicos más enigmáticos y fascinantes de la escala animal. La rápida restauración del volumen y de la función hepática tras una hepatectomía mayor (> 70 %) o un daño hepatocelular severo y su estricta regulación en el inicio y en el cese de la regeneración, es una propiedad exclusiva del hígado (1-9).
La RH es el fundamento de aplicaciones clínicas, como las resecciones hepáticas extensas (> 70 % del parénquima hepático o cinco segmentos), el trasplante segmentario de cadáver (split liver transplantation) o de donante vivo, las hepatectomías secuenciales, la embolización portal aislada o asociada a transección hepática "in situ", el soporte artificial temporal en la insuficiencia hepática aguda y de las posibles aplicaciones clínicas de la terapia celular (10-21) (Fig. 1).

Aunque la capacidad regenerativa del hígado ya estaba reflejada en las penas infligidas por Zeus a Prometeo (Eschilo: 525 a.c.) no fue hasta 1879 cuando se realizó la primera descripción científica de la regeneración hepática (22-24). Dicha capacidad representa una de las respuestas fisiológicas más extraordinarias para mantener el medio interno. Además de las funciones homeostáticas y metabólicas, el hígado es el mayor órgano sólido inmune del organismo y el primer filtro natural para micro-organismos (bacterias, virus) y moléculas potencialmente tóxicas (xenobióticos) procedentes del intestino (25-28). En los últimos 50 años ha habido un interés extraordinario en conocer los mecanismos básicos de la RH e identificar posibles aplicaciones clínicas (4,7,29-31). En el presente artículo describimos las bases fisiológicas de la RH y su relación con situaciones clínicas.
Características de la regeneración hepática
En condiciones basales los hepatocitos permanecen en estado "quiescente" o fase G0 (G cero) del ciclo celular, pero, a diferencia de otros tejidos, mantienen la capacidad de reiniciar el ciclo celular ante cualquier lesión, pérdida de tejido hepático o a estímulos exógenos (factores de crecimiento, mitógenos) (32-35).
La regeneración se ha estudiado en múltiples modelos experimentales; desde cultivos celulares, a modelos "in vivo" de hepatopatía inducidos por tóxicos (Cl4C, galactosamina, tioacetamida), productos bacterianos (LPS), virus, y en modelos quirúrgicos como la hepatectomía del 70 %, o el trasplante hepático parcial. También se ha estudiado en animales modificados genéticamente en los que se ha sobre-expresado un gen (knock-in) o en animales knock-out (KO) en los que se ha inducido la deleción específica o condicional de un gen (2,5,36-38). Otros autores han realizado estudios transcripcionales analizando la expresión de miles de genes, tratando de obtener un patrón representativo de la regeneración (39-43). En la tabla I se resumen algunos de los modelos descritos.

De los modelos referidos, destacan los que evalúan la restauración del volumen hepático, la función hepática y supervivencia de los animales. Aunque los modelos KO han sido cruciales para identificar las vías de señalización y transcripción de la RH, debido al carácter pleiomórfico y redundante de la regeneración, su interpretación es difícil (40).
El modelo más utilizado es la hepatectomía del 70 % en roedores, ya que representa el mayor estímulo de regeneración y garantiza la respuesta sincrónica y homogénea del hígado remanente (36-38). Este modelo ha permitido comparar la regeneración en animales control -"tipo silvestre"- con animales modificados genéticamente para las moléculas de señalización, sus receptores y factores reguladores del ciclo celular (2,4,5,44).
Tras una hepatectomía o una lesión aguda, la recuperación de la masa hepática se debe a una "hiperplasia compensatoria" del hígado residual, y no a una regeneración epimórfica del tejido perdido, como ocurre en vertebrados inferiores -pez Zebra o la Salamandra- los cuales la capacidad de regenerar las extremidades amputadas u otras estructuras -la cola, la mandíbula inferior, el ventrículo cardiaco- a lo largo de toda su vida (45-49).
Estos animales regeneran el tejido perdido a partir del "blastema" formado por la desdiferenciación de células adultas en células precursoras tejido-específicas y por células madre mesenquimales (45,47,48).
Las características de la RH se pueden resumir en las siguientes:
1. Se trata de un fenómeno presente en todos los vertebrados, desde el pez Zebra hasta el ser humano, del que se han descrito mecanismos comunes: participación del sistema inmune innato, desarrollo de ploidía y aneuploidía, remodelación tisular y regulación estricta del volumen hepático (34,46,50,51).
2. La RH es una respuesta controlada, según el patrón descrito en la figura 2. La duplicación de los hepatocitos, las secuencias replicativas de las células mesenquimales, la morfogénesis y el control del volumen al término de la regeneración, están regulados genéticamente. Se trata de un fenómeno distinto de la embriogénesis y de la cicatrización tisular, aunque existan vías de señalización y programas transcripcionales comunes (4,49,52).
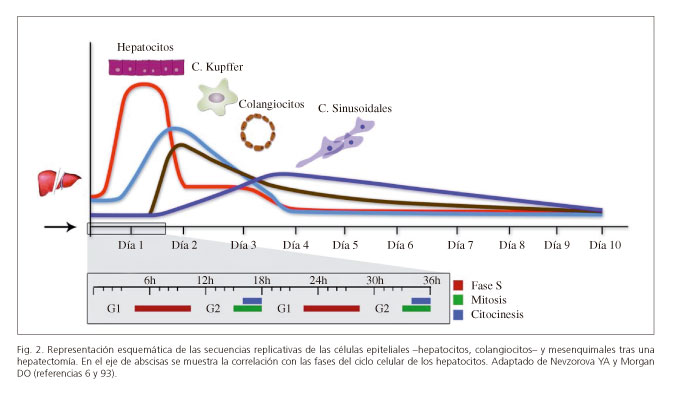
3. Es un proceso muy rápido que se inicia en segundos y termina en pocos días -5,7 días, dependiendo de la edad y de la especie-, con la restauración del volumen hepático, y en el que intervienen mecanismos de señalización -autocrinos, paracrinos y endocrinos-, los genes responsables de la proliferación celular, la morfogénesis, la "zonalización" hepática y la regulación del metabolismo (1,2,4,5,37,44,53). En los últimos años se han identificado las vías de señalización y transcripción de la RH, muchas de ellas relacionadas con la respuesta fisiológica del hígado al daño celular. En el presente trabajo veremos cómo en la RH se reproduce el patrón de los mecanismos de adaptación al daño tisular (54-56).
La RH se inicia con el reconocimiento de los patrones moleculares asociados a microorganismos patógenos (PAMP, del inglés: "pathogen-associated molecular pattern") o de los patrones moleculares asociados al daño celular (DAMP, del inglés: "damage associated molecular pattern"); generando una respuesta inmune innata -activación de las fracciones C3a y C5a del complemento, producción de TNF-α, síntesis de IL-6, IL-1β, IL-18- que estimulan la división de los hepatocitos mediante la transición del estado basal G0 a la primera fase G1 del ciclo celular (fase G1, del inglés Gap) (fase de preparación) y que ocurre en las primeras 4 horas tras una hepatectomía (57-67).
4. En la RH se reproducen las fases del ciclo celular, lo que ha suscitado un gran interés en conocer la expresión génica, los mecanismos de regulación y su traslación a otras situaciones, como el cáncer (33,35,42). La proliferación celular se sucede en un orden cronológico y secuencial; primero se dividen los hepatocitos y posteriormente las células de Kupffer y endoteliales, seguido de la neoformación de vasos y de los canalículos biliares, hasta reproducir la estructura hepática. Tras una hepatectomía del 70 %; el 95 % de los hepatocitos pasan en 4 horas de la fase G0 a la fase G1 del ciclo celular (1,4,68); al término de la cual existe el punto crítico de control, llamado de restricción ("R"), que determina si las células se dividen de forma irreversible. Hasta entonces el proceso es reversible y los hepatocitos podrían regresar a su estado previo (G0) si no se dieran las condiciones idóneas (factores de crecimiento, citocinas, etc.). Posteriormente progresan por las fases del ciclo celular hasta la división celular o citocinesis de los hepatocitos (69,70) (Fig. 2).
A los 30 minutos de una hepatectomía, se ha descrito un aumento de los factores de transcripción preformados en el citosol como respuesta a la unión de las citocinas a sus receptores: el transductor de señal y activador de la transcripción STAT3 (activador de la transcripción y transductor de la señal 3), factor nuclear Kappa-β (NF-Kβ) y la proteína activadora 1 (AP-1); que estimulan la síntesis de proteínas y la expresión de los genes necesarios para iniciar el ciclo celular (fase de preparación). En la rata, se ha descrito un incremento en la síntesis de DNA (fase S) a las 24 horas de la hepatectomía, y en el ratón, a las 36 horas. Los hepatocitos entran en mitosis de forma sincronizada a las 48 horas, seguidos de las células de Kupffer. Las células estrelladas y las del epitelio biliar tienen su fase S a las 48 horas y proliferan de forma más lenta. Las células endoteliales inician su proliferación a los 3 días, expresando un pico a los 5 días de la hepatectomía (3,4,42,68,71-73).
A las 72 horas de la hepatectomía, una subpoblación de los hepatocitos retornan al estado basal G0, mientras que otros reinician la mitosis hasta su retorno al estado previo G0, con la restauración de la masa hepática (4,6,72). Se ha postulado que este segundo pico proliferativo se debe a factores de crecimiento sintetizados por los propios hepatocitos o a co-mitógenos como la norepinefrina, insulina, somatostatina y el glucagón (1,2,4).
Recientemente, Miyaoka y cols. (52) han descrito que tras una hepatectomía del 70 %, los hepatocitos duplican su tamaño celular en las primeras 24 horas y se dividen 1,5 veces. Dichos autores han cuantificado el aumento del tamaño en 1,6 veces, y en 0,7 divisiones por célula, confirmando que la regeneración hepática se debe tanto a una hipertrofia como a la proliferación de los hepatocitos. Dichos autores observaron que, tras una hepatectomía menor (≤ 30 %), el hígado remanente aumentaba a expensas del tamaño celular en 1,4 veces; sin acompañarse de división celular (52).
Desde el punto de vista morfológico, en las primeras horas se observan acúmulos de hepatocitos (10-14 células mononucleadas y binucleadas con diferente grado de ploidía: tetraploides -4n-, octaploides -8n-; -16n-, -32n-). A los 2-4 días, se observa la proliferación de células estrelladas y sinusoidales, aumentando el tamaño de los lóbulos (35,74,75).
Ding y cols. (76) han confirmado la secreción de factores angiocrinos y la neoformación de vasos. La hipoxia relativa del hígado remanente induce la expresión de factor de transcripción, inducido por hipoxia (HIF-1α) -a las 12-48 horas de la hepatectomía parcial- que activa el factor de crecimiento endotelio-vascular (VEGF), el factor de crecimiento fibroblasto (FGF) y la sintetasa inducible de óxido nítrico (iNOS) (77-81). La proliferación de los hepatocitos progresa desde las áreas periportales a las áreas pericentrales del lobulillo. Una vez re-establecido el volumen inicial se produce un fenómeno de apoptosis, remodelación tisular y zonalización de los hepatocitos (42,44,72,82,83).
5. A diferencia de otros órganos sólidos como la piel o el intestino, la RH se debe a la proliferación de los hepatocitos maduros y de las células mesenquimales: células de Kupffer, endoteliales, estrelladas, linfocitos NK, y no a la proliferación y diferenciación de las células madre pluripotenciales o a las células ovales; si bien se ha descrito su proliferación cuando la regeneración hepática es insuficiente o está abolida, como en hepatopatías crónicas (1,2,3,5,42,44,72,84-87).
Con la identificación de las células ovales -precursoras de los hepatocitos y colangiocitos-, así como el descubrimiento de la plasticidad tisular de las células madre hematopoyéticas, de las células progenitoras endoteliales (CD133, CD117) y de las células madre pluripotenciales inducidas (iPS), descritas en 2005 por el premio Nobel de 2012 Shinya Yamanaka (88-90), en los últimos 20 años se ha producido una explosión de trabajos relacionados con la terapia celular en todo tipo de hepatopatías (91,92). Debido al ingente número de trabajos, referimos al lector a unas revisiones excelentes (93-97). A pesar de las enormes expectativas, los resultados han sido escasos incluso en los ensayos realizados con hepatocitos aislados en los errores congénitos del metabolismo, limitándose en la mayoría de los casos a una mejoría temporal (20,98). Recientemente se han publicado estudios preliminares mediante la infusión de factores de crecimiento de médula ósea (factor estimulante de granulocitos; G-CSF) en pacientes con el síndrome de insuficiencia aguda sobre crónica, y con la infusión de células progenitoras de médula ósea (CD133) en hepatopatías crónicas con fines regenerativos (96-99). Los resultados clínicos, al igual que ha ocurrido en otros órganos sólidos (corazón, páncreas, intestino), sugieren que la regeneración tisular es un proceso más complejo y dependiente de la cooperación específica entre diferentes estirpes celulares (87,100-102).
6. La capacidad regenerativa del hígado es casi ilimitada. Se han descrito hasta 7 hepatectomías del 50 % consecutivas en la rata, sin provocar insuficiencia hepática ni disminuir su potencial regenerativo (2-4). En un modelo de tirosinemia homozigota en el ratón (FAH -/- Miles), el trasplante de hepatocitos corrigió el déficit metabólico. Cuando se aislaron los hepatocitos trasplantados, estos revirtieron el déficit enzimático en una segunda generación de ratones FAH -/-; y así sucesivamente hasta ¡diez generaciones! Se ha estimado que los hepatocitos de un ratón podrían sufrir hasta 69 duplicaciones (generar 50 hígados de ratón) (103,104).
7. Otro enigma de la RH es la regulación estricta entre el volumen hepático y la superficie corporal, expresado como índice hepático (peso hígado/peso total x 100 ~ 2,5 %), conocida como regulación "hepatoestática". En el trasplante clínico y experimental -injertos pequeños en receptores grandes y viceversa- se ha confirmado la adaptación del volumen hepático donante a la superficie corporal del receptor. Dicho control se ha descrito incluso en el xenotrasplante de babuino al hombre (4,105-107). Aunque la regeneración se ha relacionado con los cambios en el núcleo, durante la división celular se deben duplicar los orgánulos citoplasmáticos. El control de la relación entre el tamaño del citoplasma y la duplicación de los cromosomas es uno de los mecanismos peor conocidos (69,108-110). La mayoría de los factores de crecimiento (el PDGF, el factor de crecimiento epidérmico -EGF-, el factor de crecimiento hepatocitario -HGF- y el factor de crecimiento insulínico -IGF-1-), además de tener efectos mitógenos, aumentan el tamaño y la supervivencia celular, inhibiendo la apoptosis (5,69,108). En los vertebrados inferiores y en los mamíferos, es frecuente observar hepatocitos poliploides, tetraploides (4n), octaploides (8n), bien mononucleados o binucleados (2 x 2n; 2 x 4n; 2 x 8n) a consecuencia de una alteración en la citocinesis (Figs. 3 y 4). El grado de ploidía varía según las especies; en las ratas el 80 % de los hepatocitos son poliploides y aneuploides, en el ratón el 60 % y en el ser humano entre 30-70 % (75,111). La primera consecuencia de la ploidía es el aumento del tamaño de los hepatocitos (2,112,113). Se ha postulado que el tamaño celular depende del contenido nuclear de DNA, de forma que un número mayor de cromosomas permite aumentar el tamaño (114-116).


Tras una hepatectomía en roedores se ha descrito una disminución de los hepatocitos binucleados desde el 20 % al 5 % y un aumento de los hepatocitos tetraploides -4n- y octaploides -8n-; e incluso hepatocitos 16n a las 72 horas, expresando que la hepatectomía es un estímulo muy intenso para la duplicación del DNA previo a la citocinesis (2,112,116,117).
En animales en los que se había inhibido la separación de los cromosomas, la recuperación de la masa hepática tras una hepatectomía se debió a la poliploidía de los hepatocitos -18n, 32n y n superiores- (118,119). Este fenómeno -conocido como endoreduplicación- da lugar a múltiples secuencias replicativas de los cromosomas sin la división celular, generando células con varias copias del genoma, capaces de aumentar la expresión génica (120). Duncan y cols. (35,74,111) han descrito la capacidad de los hepatocitos para desarrollar poliploidía y su reversión incluso a la aneuploidía como mecanismo de adaptación a estímulos xenobióticos o dietéticos. Es llamativo que en estos casos la función hepática fuera normal, confirmando la equivalencia funcional entre hepatocitos 2n, 4n y 8n (2,112).
Recientemente Kurina y cols. (117) han descrito el papel regulador de p53 en la integridad del genoma, tanto en hepatocitos quiescentes como durante la regeneración (7 días post-hepatectomía). Dichos autores han descrito que los ratones KO para p53 (p53 -/-) expresan un grado mayor de ploidía que ratones control p53 +/+, y que inician antes la división, desarrollan un tamaño mayor del núcleo y del citoplasma, y un grado mayor de ploidía que ratones p53 +/+ (tipo silvestre o control) tras una hepatectomía del 70 %. La ausencia de p53 altera la reversión de la ploidía, por lo que la regeneración en animales p53 -/- se debe primordialmente al aumento del tamaño celular. Aunque la recuperación de la masa hepática a los 7 días de la hepatectomía era equivalente en ambos grupos de ratones, dichos autores observaron más errores en la integridad cromosómica en los ratones p53 -/- (husos multipolares y cromosomas perezosos) que en los animales control (p53 +/+) (52,117,121,122).
8. Diversos autores han resaltado que cuanto más severo es el daño hepático (resecciones extremas > 70 % del parénquima, trasplante entre vivos con injertos ≤ 30 %, necrosis hepática submasiva...), más intensa es la respuesta proliferativa del hígado, y viceversa (52,113-116). En el trasplante entre vivos se ha descrito que la regeneración del injerto en el receptor es más rápida que la del hígado remanente en el donante, y que los injertos más pequeños (≤ 30 % del volumen estándar del hígado) regeneraban más rápidamente que los injertos mayores (≥ 40 %, considerado este como el mínimo aceptable) (123,124). Este patrón es similar a la respuesta fisiológica al estrés o al daño tisular (32,125-132).
9. El hígado -a pesar de su complejidad metabólica- mantiene las funciones homeostáticas: síntesis de proteínas, factores de coagulación, anti-proteasas, detoxificación de xenobióticos, etc. durante la regeneración. Estudios con microarrays han mostrado una respuesta selectiva en las primeras 40 horas, priorizando la expresión de los genes relacionados con la división celular y silencionando los relacionados con el metabolismo hidrocarbonado (42,72). Estudios con microscopia óptica y electrónica han revelado, a las 24 horas de una hepatectomía del 70 %, cambios selectivos en los hepatocitos de la zona 1, en contraste con la zona 3 del lobulillo hepático (34,42,72).
Wu y cols. (82) han referido que existen tres picos en la síntesis de DNA de los hepatocitos, inicialmente en la zona 1 y posteriormente en la zona intermedia del lobulillo. Curiosamente el 15 % de los hepatocitos no se dividen, mientras que un 11 % de los hepatocitos se dividen al menos tres veces, desconociéndose la causa.
10. El hígado es, con el cerebro, el órgano con mayor capacidad de mantener su integridad -morfológica y funcional- y responder a las alteraciones más variadas del medio interno. Esta propiedad está presente en otros "sistemas biológicos": homeostasis fisiológica, desarrollo de los tejidos, ciclo celular, resistencia ecológica, ritmo circadiano e incluso en el cáncer (133-135). Dicha propiedad se ha denominado como "robustness" o "fortaleza" y ha despertado gran interés en la biología evolutiva durante la última década. La RH reúne las cualidades requeridas: pleiomorfismo, redundancia y mecanismos de bio-feedback. Esto hace que su comprensión sea difícil y que, todavía, no se haya podido intervenir en la regeneración con fines terapéuticos (2,4,10,44).
Fases de la regeneración hepática
Se debe a Fausto y cols. (1,2) el mérito de haber integrado en un modelo "trifásico" los mecanismos celulares y moleculares de la RH. La RH puede dividirse en tres fases: una fase inicial de "preparación" o "cebado", que corresponde al paso de los hepatocitos quiescentes de la fase G0 a la fase G1 del ciclo celular, que ocurre en las primeras 4 horas tras la hepatectomía. Una segunda fase, o fase de "progresión", que corresponde a la transición desde la fase G1 hasta completar la mitosis (citocinesis); y una tercera fase de apoptosis, remodelación tisular y retorno a la fase G0. El modelo descrito se ha confirmado en especies diferentes y tiene la virtud de corresponderse con las fases del ciclo celular y del que se describen algunos aspectos básicos (32,69,136-138).
El ciclo celular se divide en cuatro fases: G1, S, G2 y M, según se describe en la figura 5. La síntesis del DNA, y la duplicación de los cromosomas, ocurre en la fase S (de síntesis). La segregración de los cromosomas tiene lugar en la fase M o mitosis, que se divide a su vez en cuatro etapas: profase, metafase, anafase y telofase.

La división celular termina con la citocinesis, dando lugar a dos células semejantes que pueden reiniciar el ciclo o volver al estado de reposo G0. Las fases G1 y G2 (del inglés "Gap") o fases de "espaciado" o "preparativas" situadas antes de la fase S (síntesis de DNA) y de la fase M (mitosis) respectivamente, proporcionan tiempo para el crecimiento celular y regulan la transición a la fase siguiente dependiendo de señales intracelulares y extracelulares (69,137-139).
En el ciclo existen tres puntos de control: el punto "R" o de restricción que regula la entrada en el ciclo al final de la fase G1; y los dos puntos de control de la mitosis; el punto G2/M que controla el inicio de la mitosis y el punto de control de la transición metafase-anafase que da lugar a la segregación de las cromátidas "hermanas" y al término de la mitosis (69,70,136,140-142). El punto "R" regula la velocidad de la división celular y determina la irreversibilidad del ciclo. Una vez traspasado dicho punto la célula se dividirá inexorablemente aunque continúe sometida a los mecanismos de control (69).
La mayoría de los mitógenos y de los factores de crecimiento -factor de crecimiento derivado de plaquetas (PDEGF), el EGF, el HGF y el factor de crecimiento transformador β (TGFβ)-, ejercen su acción en el punto de control "R" cuando la célula es más sensible a factores exógenos (138-140).
Fase inicial de "preparación" de los hepatocitos
Respuesta inicial inflamatoria estéril
Según el modelo descrito, la regeneración hepática es una respuesta fisiológica de adaptación al daño hepatocelular (63,143). El daño tisular se puede manifestar como "estrés celular" o como una de las formas de muerte celular: necrosis, apoptosis o necro-apoptosis (58,144-147). La necrosis induce la respuesta del sistema inmune innato debido al reconocimiento de los DAMP por los receptores de reconocimiento de dichos patrones (RRP). Dichos receptores se han descrito en la membrana plasmática, en las membranas endosómicas y en el citoplasma de las células mesenquimales y de los hepatocitos (44,54,55,61,62,148,149).
Dependiendo de la severidad de la lesión y de la respuesta regenerativa, la lesión será reversible o irreversible. En este último caso el hígado es incapaz de restaurar la homeostasis, lo que se asocia a una mortalidad muy elevada y cuya única alternativa es el trasplante hepático (hepatitis fulminante, síndrome "small-for-size", insuficiencia hepática aguda sobre crónica, insuficiencia hepática post-hepatectomía) (150-152). En dichas situaciones se desarrolla un cuadro de colestasis progresiva, coagulopatía, encefalopatía, sepsis y fallo multiorgánico; que algunos autores han relacionado con una respuesta inmune insuficiente. Otros autores han sugerido que un estímulo "mitogénico" excesivo, generaría el fenómeno conocido como "respuesta al estrés hiperproliferativo", que induce la apoptosis celular (153-155).
En 1994 Polly Matzinger formuló la teoría denominada "Danger", por la que el sistema natural inmune, además de reconocer los gérmenes patógenos y los componentes de la pared bacteriana, reconoce y responde a ligandos endógenos derivados del daño celular y de la necrosis, lo que se ha llamado "respuesta inflamatoria estéril" (54,55,59,156-158).
En la necrosis, el paso de moléculas intracelulares -DAMP- al espacio extracelular, estimula a los receptores de dichos patrones (RRP). Entre dichos receptores se encuentran el sistema del complemento y los receptores tipo Toll (RTT; del inglés "toll-like receptors") que se expresan en las membranas y en el citosol de los macrófagos, las células endoteliales, las células dendríticas, células NK y de los hepatocitos (159-163) (Fig. 6).

Los DAMP también son reconocidos por una subfamilia de receptores citoplásmáticos del daño y del estrés celular, denominados NLRP y pertenecientes a los receptores tipo-NOD (NLRs, NOD-like receptors) que una vez estimulados forman el complejo denominado "inflamosoma" y activan la liberación de interleucina-1β (IL-β) y de la interleucina-18 (IL-18), con efectos inflamatorios y promitóticos. El papel del receptor NLRP en la inflamación y en los mecanismos de supervivencia, ha despertado un interés extraordinario en los últimos años (164-166).
Desde la formulación de la teoría Danger por Matzinger, se han identificado DAMP relacionados con el daño celular: proteína de alta movilidad del grupo caja 1 (HMGB1), proteínas del choque térmico (Hsp, Hsp60, Hsp70), ácido úrico, DNA genómico, ATP, adenosina, heparan sulfato, oligosacáridos, fragmentos de degradación del ácido hialurónico, péptidos N-formilados mitocondriales, y que se han denominado con el nombre genérico de "alarminas" (59,63,167-169).
Es significativo que las proteínas de reconocimiento del sistema innato sean filogenéticamente anteriores a la separación entre animales y vegetales (hace mil millones de años) y a la adquisición del sistema inmunitario adaptativo. Desde la descripción de los receptores RTT en la Drosophila y en el ser humano, y de la teoría DANGER, se ha descrito el paradigma de la respuesta inmune innata en la reparación y regeneración tisular (170-176).
La unión de los receptores tipo Toll con sus ligandos, estimula varias cascadas de señalización que culminan con la expresión de IL-6, TNF-α e IL-β, y la activación de factores de transcripción citosólicos -NF-kβ, STAT-3, AP-1- que se traslocan al núcleo y ejercen efectos proliferativos (62). Los RTT participan en la regeneración mediante señales de pro-supervivencia e inhibición de la apoptosis en la mucosa intestinal en el colon, pulmón, piel y en la regeneración hepática tras la hepatectomía (63,170,177-179).
La RH también se ha relacionado con ligandos endógenos presentes en la circulación enterohepática: ácidos biliares (Abs), xenobióticos, LPS y con componentes de la matriz extracelular -fibronectina, heparan sulfato, fibrinógeno, oligosacáridos del ácido hialurónico- derivados de la lesión tisular (maniobras quirúrgicas) y diferentes tipos de estrés (180-185).
Las células mesenquimales, especialmente los macrófagos, una vez activadas por los DAMP, sintetizan citocinas proinflamatorias como la IL-1β; el TNF-α, la IL-6, el interferón-gamma (IFN-γ), prostaglandinas y el factor activador de las plaquetas (PAF) con funciones proliferativas y antiapoptóticas (63,167,186-188).
Las citocinas TNF-α e IL-6 inician la fase de preparación o "cebado" de los hepatocitos (transición G0-G1 del ciclo celular). Dichas citocinas señalizan a través de receptores para TNF (TNF-RI, TNF-RII) y para IL-6 (IL-6R/gp130). Son receptores tirosina-quinasas similares a los de los factores de crecimiento, que activan cascadas enzimáticas como las quinasas activadas por mitógenos (MAPK, mitogen activated protein kinases). A este grupo de quinasas pertenece la quinasa Janus, conocida como JAK, que una vez activada fosforila el factor de transcripción preformado STAT-3 (activador de la transcripción y transductor de la señal 3) (señalización JAK/STAT). La proteína STAT-3 se une al DNA promoviendo la expresión de los genes de respuesta precoz inmediata (IEGs): c-fos, c-jun y c-myc (llamados oncogenes), responsables del inicio del ciclo celular (Fig. 6). Se han descrito más de 180 de dichos genes que sintetizan las proteínas necesarias para abandonar el estado basal G0 (1,4,5,42,44,72,118,188-191).
Esta fase de preparación o "cebado" se inicia en los primeros 30 min y perdura durante las primeras 4 horas post-hepatectomía. Además del factor STAT-3, otros factores de transcripción preformados son el factor nuclear Kappa-β (NF-kβ) y la proteína activadora 1 (AP-1); necesarios para la síntesis "de novo" de las proteínas reguladoras de la transición G0-G1 y G1/S. Con técnicas de manipulación genética en animales knock-out y knock-down se ha confirmado la participación de STAT-3, NF-kβ y AP-1 en la regeneración hepática (192-195).
Animales con mutaciones en los receptores de TNF-α (TNF-R1), expresaban una inhibición de la transcripción de NF-kβ y una alteración severa de la regeneración, confirmando que la transducción de NF-kβ en las células de Kupffer es crucial en la respuesta a las citocinas. Ratones KO para los receptores gp130 de la IL-6 mostraban defectos menores en la proliferación celular. En animales KO para IL-6 (IL6 -/-) y su receptor (gp130), la administración de LPS tras una hepatectomía redujo la supervivencia, evidenciando el papel protector y estimulador de la IL-6 en la regeneración. La deleción hepato-específica de STAT-3 y de AP-1 disminuye la expresión de las ciclinas, cruciales para la transición de G0-G1 y G1-S del ciclo celular. Por el contrario, en animales en los que se indujo un aumento del factor NF-kβ -mediante el bloqueo de su inhibidor citoplasmático IkbKβ- se observó una respuesta inflamatoria y proliferativa más intensa (72,196).
Además de los mediadores inflamatorios -TNF-α, IL-6, C3a, C3b, LPS-, los factores de crecimiento como el HGF, PDGF y EGF también estimulan el paso G0-G1. El HGF señaliza a través de su receptor c-met, activa a STAT-3 e induce la expresión de genes de respuesta precoz inmediata. Ratones con una mutación condicional en el receptor Met, expresan un defecto en la regeneración (6,197,198).
Como veremos en la fase de proliferación, el HGF también regula la expresión de genes relacionados con otras fases del ciclo celular y tiene efectos de "pro-supervivencia" mediante la inhibición de la apoptosis (4).
Sistema del complemento y regeneración hepática
En modelos murinos de daño hepático -Cl4C, hepatectomía parcial- y en pacientes sometidos a una hepatectomía, se ha descrito un aumento de las fracciones activadas del complemento C3a y C5a en las primeras 24 horas (131,132,149). En ratones deficientes (KO) en la fracción C3 (C3 -/-) y en la fracción C5 (C5 -/-), se observó una alteración en la regeneración tras una hepatectomía del 70 %. En dichos animales se observó una reducción en los niveles de TNF-α y del ARNm de IL-6, y una disminución en los factores de transcripción NF-kβ y STAT-3 (172-174). La administración de un antagonista del receptor de C5a (C5a R) tuvo efectos similares con un aumento de la mortalidad; confirmando el papel del complemento en la fase inicial de cebado de los hepatocitos (199).
Los animales KO, C3 -/- y C5 -/-, desarrollaron lesiones hepáticas más severas que los animales control tras el daño inducido por la infusión de Cl4C y tras una hepatectomía parcial. En dichos animales los defectos en la regeneración y en el daño hepatocelular revirtieron tras la reconstitución con C3a y C5a. El mismo fenómeno y más intenso se observó en animales con la doble mutación (C3/C5 -/-) así como la recuperación de la regeneración con la administración de C3 y C5.
El grupo de Lambris (170) ha descrito que la fracción C3a induce la síntesis de IL-4 por las NKT en las primeras 24 horas de la hepatectomía y estimula la síntesis de las proteínas del complemento y de la IL-6 por los macrófagos en la fase de cebado de los hepatocitos. Las funciones reparadoras y tróficas del sistema del complemento han suscitado gran interés en la regeneración tisular. El sistema del complemento, además de su acción frente a bacterias, virus, hongos y células tumorales, es el componente humoral más rápido para reconocer el daño tisular a través de los DAMP e iniciar la respuesta de reparación tisular (170,199).
Plaquetas y regeneración hepática
Además de las funciones hemostáticas, las plaquetas expresan receptores tipo Toll 2, 4 y 9 y participan en la respuesta inmune innata. Dichos receptores reconocen los PAMP y los DAMP, por lo que algunos autores las consideran como los "guardianes circulantes" del daño tisular y un vestigio de los hemocitos de los invertebrados (200,201).
Las plaquetas contienen, en sus gránulos, fibrinógeno, factor Von Willebrand, proteínas de adhesión, factores proangiogénicos con efectos mitógenos, hepatoprotectores (VEGF, PDGF, HGF, IGF, EGF-1 y TGFβ), y el ¡95 %! de la serotonina circulante. Aunque son células anucleadas tienen ARN y pueden sintetizar más de 300 proteínas diferentes entre las que destaca el TGFβ (202,203).
En 1996 Tomikawa y cols. (204) describieron que la trombocitosis secundaria a la esplenectomía, estimulaba la regeneración hepática en roedores. Estudios posteriores confirmaron dichos resultados y el efecto opuesto de la trombocitopenia. El mismo grupo describió un aumento de la supervivencia mediante la inducción de trombocitosis en un modelo de hepatectomía subletal (> 90 %). Otros grupos han relacionado las plaquetas con un efecto protector sobre la disfunción hepática post-hepatectomía, e incluso con la mortalidad operatoria cuando eran < 100.000 plaquetas/μl. Los autores relacionaron estos resultados con los factores de crecimiento citados y el factor de crecimiento insulínico (IGF-1) (205,206).
Lesurtel, describió en 2006 el efecto estimulador de la serotonina en la regeneración hepática "in vivo", aunque ya se conocían sus efectos "mitógenos" "in vitro" sobre fibroblastos y hepatocitos (207). El mismo grupo confirmó sus hallazgos en animales KO para la enzima precursora de la serotonina (triptófano hidrolasa 1) (TPM1 -/-) y han descrito un aumento de la supervivencia en un modelo murino de "small-for-size" con la administración de un agonista sintético de la serotonina; 5-HT2B. En los animales tratados observaron un aumento en las fenestraciones endoteliales de los injertos (208).
Estos autores han descrito la reversión de la "pseudocapilarización" de los sinusoides en animales "viejos" y un aumento de la supervivencia tras una hepatectomía con la administración del mismo agonista; sugiriendo que la apertura de las fenestraciones endotelio-sinusoidales facilitaría el contacto directo de las plaquetas, citocinas y de los factores de crecimiento con los hepatocitos (208). Otros autores han atribuido el efecto proliferativo de la serotonina con el estímulo en la síntesis de VEGF. Por el contrario, Matondo y cols. (209) en un modelo murino KO (TPH1 -/-) deficiente en el transportador de membrana de la serotonina no observó ningún efecto tras una hepatectomía del 70 %.
Citocinas, factores de crecimiento y hormonas
En 1967 Moolten y Bucher describieron que el plasma de ratas hepatectomizadas tenían efectos mitógenos sobre los hepatocitos en modelos "in vitro" e "in vivo". Desde entonces se han identificado citocinas, factores de crecimiento, hormonas y metabolitos relacionados con el ciclo celular de los hepatocitos y de las células mesenquimales (4,210-213).
Citocinas de la familia IL-6
La IL-6 es una interleucina tipo I, asociada con la respuesta inmune innata. Debido a sus efectos citoprotectores, proliferativos y antiapoptóticos es la más relacionada con la regeneración hepática. Posteriormente se aislaron otras 7 interleucinas muy similares a la IL-6, que comparten los receptores de membrana tirosina-quinasa gp80 y gp130 como vía de señalización y que se han englobado con el término "familia de IL-6": IL-6, interleucina 11, factor inhibidor de leucemia (LIF), oncostatina (OSM), factor neurotrófico ciliar (CNTF), cadriotrofina (CT-1) y la IL-27 (186-188,214-217).
La IL-6 se libera precozmente por los macrófagos como respuesta al daño tisular. Tiene efectos locales sobre los hepatocitos y sistémicos -fiebre, somnolencia, secreción de ACTH y vasopresina- propios de la respuesta sistémica al estrés. Estimula la síntesis hepática de los reactantes de fase aguda (proteína C reactiva, amiloide A y además tiene efectos proinflamatorios similares al TNF-α e IL-1 (218-221).
La IL-6 se une a receptores de membrana gp130, provocando la dimerización de dos subunidades gp130 (222). Dichos receptores están unidos a las tirosina-quinasas que residen en el citoplasma, denominadas tipo Janus (JAK1, JAK2, JAK3 y TyK2). La dimerización de gp130 y su unión con el receptor correspondiente (IL-Rα) promueve la activación de las quinasas tipo JAK que fosforilan y activan los factores de transcripción como STAT-3. Las proteínas STAT (7 en el hombre), ubicadas en el citosol, migran al núcleo para unirse a promotores del DNA y estimular la transcripción génica de la proliferación celular, supervivencia y/o muerte celular (Fig. 6) (223,224).
El STAT-3 activado induce la transcripción de los genes de respuesta precoz inmediata c-jun, c-myc, c-fos y el de crecimiento precoz (Egr-1). Además, la vía IL-6 / JAK-STAT-3 participa en la síntesis de proteínas antiapoptóticas Bcl-2 y Bcl-x. La vía JAK-STAT es una de las vías de transducción de señales más directas de la membrana al núcleo. La deleción hepatoespecífica de STAT-3 provoca una disminución en la expresión de las ciclinas D1 y E1, necesarias para iniciar el ciclo celular (72,191,192).
Además de STAT-3 existen otras dos vías de transcripción mediadas por receptores tirosin-quinasa, el factor nuclear KB (NF-kβ) y la proteína activadora 1 (AP1). El factor NF-kβ se encuentra habitualmente inactivo en el citoplasma debido a la proteína inhibidora IkbKβ. La unión de una citocina con su receptor estimula la degradacion del inhibidor IkbKβ, liberando el factor NFkβ que se trasloca al núcleo y activa la transcripción de las proteínas de la fase aguda y los genes pro-proliferativos y antiapoptóticos mencionados. Tras una hepatectomía (70 %) o un daño hepático -isquemia, Cl4C, administración de LPS- se produce una rápida elevación de TNFα e IL-6. Ratones KO para IL-6 (IL-6 -/-) tras una hepatectomía desarrollaron un fallo hepático que revertía cuando previamente se administraba IL-6 (225). En los animales IL-6 -/- apenas se inducía la activación de STAT-3. En modelos KO condicionales para STAT-3 (ya que el STAT-3 -/- es letal en el periodo embrionario) se ha confirmado su efecto protector en lesiones inducidas por adenovirus (198). En modelos murinos de hepatotoxicidad, de isquemia-reperfusión y colestasis severa, se ha descrito el efecto citoprotector de la IL-6. Recientemente se ha confirmado el efecto antiapoptótico de la IL-6 en hepatectomías extremas (87 %) y la rápida regeneración de injertos "pequeños" (≤ 30 %) evitando el síndrome "small for size" (226-229). El factor de transcripción AP-1 promueve la expresión de los genes de respuesta precoz inmediata en las primeras 5 horas post-hepatectomía; especialmente la oncoproteína Jun que facilita la transición G0-G1 y G1-S (72).
Una de las citocinas de la familia IL-6 que ha suscitado gran interés es la cardiotrofina-1 (CT-1). El grupo de Prieto y cols. ha descrito su efecto protector en la apoptosis e inductor de la regeneración en modelos de isquemia-reperfusión, hepatectomía extensa (92 %) y de hepatitis fulminante (230-232).
Receptores de vías metabólicas y xenobióticas
Además de la capacidad de los hepatocitos para pasar del estado quiescente (G0) al de división celular, el hígado remanente debe realizar las funciones metabólicas específicas del hígado durante la regeneración (2,4,5,42,72). La expresión de los genes relacionados con el metabolismo son inicialmente suplantados por los genes relacionados con la formación del citoesqueleto, el ensamblado del huso mitótico y la mitosis. Estudios transcripcionales han mostrado que en las primeras horas se produce un silenciamiento de los genes metabólicos (primeras 40 horas tras la hepatectomía) y una recuperación al término de la mitosis (42,233).
Se ha confirmado que los mecanismos de transcripción dependientes del metabolismo de los ácidos biliares (Abs), de la detoxificación de drogas y de la regulación del metabolismo hidrocarbonado, participan en la fase inicial de preparación o "cebado". Es bien conocida la regulación hepática del "pool" de los ácidos biliares en la circulación enterohepática (234). En humanos este "pool" se mantiene entre 2 y 4 g, y recirculan unas 12 veces al día, y en ratones se mantiene en los 4 mg (235). Un aumento de los ácidos biliares daña las membranas celulares, provoca un daño mitocondrial y puede conducir a la apoptosis y necrosis celular (236,237).
La homeostasis de los ácidos biliares se debe a receptores nucleares, entre los que se encuentra el "Farnesoid X-Receptor" (FXR). La unión de los ácidos biliares libres y conjugados con el dominio de unión de FXR, estimulan la transcripción de factores involucrados en las fases precoces de la regeneración (G1-S), como el FoxM1b, y de genes proliferativos (Cdc25) (166,180).
Al realizar una hepatectomía del 70 %, se provoca un aumento brusco del flujo portal al hígado remanente (1/3 de la masa original) por lo que se triplica el aporte de nutrientes y ácidos biliares procedentes de la circulación esplácnica (2,3,4,180,238-240).
Ratones deficientes en el receptor FXR muestran un retraso en la regeneración hepática y un aumento en la mortalidad tras la hepatectomía parcial (180). Estos animales son incapaces de activar los genes de respuesta inmediata c-myc, c-fos y c-jun. Por el contrario, en ratones sometidos a una hepatectomía parcial, la administración de ácidos biliares estimulaba la RH, y la administración de colestiramina retardó dicho proceso.
Los ácidos biliares estimulan la secreción de citocinas proinflamatorias como el TNF-α y la IL-1β por las células de Kupffer, con los efectos de prosupervivencia, antiapoptóticos y proliferativos (240). Por lo tanto, los ácidos biliares y los xenobióticos podrían comportarse como moléculas DANGER estimulando el inicio de la fase de preparación, además de los efectos mitógenos ya comentados sobre los hepatocitos, especialmente los de naturaleza más hidrofóbica.
Se ha descrito que la administración de agonistas del receptor FXR estimula la regeneración en animales añosos. Estos hallazgos, junto con los efectos citoprotectores de dichas moléculas, han sugerido su posible aplicación clínica (241,242).
La relación de las vías metabólicas con la regeneración es compleja, puesto que las variaciones metabólicas descritas pueden ser un epifenómeno secundario al bloqueo precoz de los genes "metabólicos", sin representar un estímulo regenerativo per se. Por ejemplo, algunos autores han descrito que la hipoglucemia persistente en ratones tras una hepatectomía estimula la regeneración, mientras que la adición de glucosa tras una hepatectomía parcial inhibe la regeneración (42,243). Dichos hallazgos parecen contradictorios con lo reseñado por Fisette y cols. (244,245); la infusión previa de glucosa e insulina en pacientes sometidos a una hepatectomía mayor, mejoraba la disfunción hepática y la regeneración.
La capacidad del hígado para responder a estímulos endógenos -ácidos biliares- y exógenos -xenobióticos-, al margen de los estímulos mitogénicos de las citocinas y factores de crecimiento, es exclusivo del hígado y puede explicar su potencial casi ilimitado de respuesta "robusta" a estímulos tan diversos como una hepatectomía, isquemia o una sobrecarga de ácidos biliares (4,42,240).
Fase de progresión
Transducción de señal mitogénica
Aunque no existe un límite nítido entre las fases de la RH, la fase de progresión comprende desde la fase G1 hasta la división celular (2,4,5,9). En este periodo, además de la cromatina, se deben duplicar los orgánulos citoplasmáticos -mitocondrias, aparato de Golgi, lisosomas, retículo endoplásmico- necesarios para preservar la función hepática y mantener una relación constante entre el tamaño del citoplasma y del núcleo (69,73,109,110).
La fase de progresión está regulada por los factores de crecimiento ya mecionados, el HGF, EGF, VEGF y TGF-α, que además de efectos mitógenos, tienen efectos tróficos y de pro-supervivencia (antiapoptóticos). La función primordial de los factores de crecimiento es activar los complejos ciclina-Cdks que inician y promueven los cambios del ciclo celular: duplicación del huso mitótico y la replicación del DNA en la fase S.
De forma similar a las citocinas, la mayoría de los factores de crecimiento también transmiten la señal mitógena al núcleo a través de los receptores de membrana y citoplasmáticos con actividad tirosina-quinasa (RTK) que promueven la cascada de las quinasas. La cascada MAP-quinasa induce la expresión de los "genes de respuesta precoz inmediata propoliferativos" (c-fos, c-jun, c-myc) cuya expresión es el primer evento transcripcional (dos horas tras una hepatectomía). Entre los factores codificados por estos genes promitóticos destacan los factores FosM1b y Myc (1-6,42,43,246).
El factor FosM1b aumenta durante la primera hora post-hepatectomía y facilita el ensamblado y activación del factor de transcripción AP-1, que estimula la expresión de los "genes de respuesta tardía" (dos días post-hepatectomía). Estos genes también codifican las ciclinas de la fase G1 (D1, D2, D3) necesarias para el inicio del ciclo (42).
Los ratones KO para c-jun, fallecen el 50 % tras una hepatectomía. El déficit en la regeneración se asocia a un incremento en la muerte celular y a un acúmulo de lípidos en los hepatocitos. A nivel molecular se observa un aumento de la proteína inhibidora de la transición G1-S.
El factor FosM1b promueve la expresión de los genes de respuesta tardía (al segundo día post-hepatectomía) y la transición G2-M. Estudios en ratones KO hepato-específicos para FosM1b, han confirmado el papel de dicho factor para la entrada en la mitosis y segregación cromosómica (247). Se ha observado que hepatocitos que sobreexpresaban FosM1b "reconstituían" los hígados de ratón previamente dañados, más eficientemente que hepatocitos de animales control. Este efecto se confirmó incluso en hepatocitos "añosos", albergando la posibilidad terapéutica en pacientes mayores con hepatopatías crónicas (248).
El factor Myc permanece elevado durante todo el ciclo e induce la transición por el punto de control "R" y de los genes relacionados con el tamaño celular y el metabolismo (108). Los niveles de ciclina D también permanecen elevados durante todo el ciclo mientras el mitógeno esté presente, sugiriendo que la expresión de la ciclina D responde al estímulo mitogénico con independencia de la fase del ciclo (108).
En las células quiescentes -fase G0- los escasos complejos ciclina D-CdK están inhibidos mediante la fosforilación de la ciclina D lo que genera su exportación fuera del núcleo y su destrucción en el citoplasma. Dicha función está catalizada por la quinasa glucógeno sintetasa (GSK3β), muy activa en los hepatocitos en reposo. La GSK3β también inhibe otros factores que promueven la proliferación celular, como la expresión génica de la ciclina D1, del factor AP-1, de Myc, y la expresión de la ciclina D2 y Cdk4. La GSK3β fosforila y destruye la b-catenina citoplasmática, bloqueando su traslación al núcleo e inhibiendo su estímulo proliferativo (108).
Los factores de crecimiento y hormonas como la insulina que señalizan a través de la cascada de las quinasas Ras -MAP- fosforilan e inhiben la GSK3β, permitiendo la síntesis de glucógeno y la activación de los factores preformados AP-1 y Myc que estimulan la proliferación celular.
Hassanain y cols. (244,245) han descrito el efecto beneficioso de la infusión de insulina y dextrosa en pacientes sometidos a una hepatectomía mayor. Dichos autores observaron un aumento del glucógeno hepático y una mejora de la función hepática postoperatoria. Además del efecto protector del glucógeno como fuente de energía para la división celular, la inhibición de la GSK3β facilitaría la expresión génica necesaria para la respuesta regenerativa. El efecto trófico de las hormonas pancreáticas sobre el parénquima hepático y la regeneración ya fue descrito por Starzl y cols. (249-252) en 1967.
Factor de crecimiento de los hepatocitos (HGF)
El HGF se aisló en 1984 al identificar el factor sérico de ratas hepatectomizadas que estimulaba la proliferación de hepatocitos in vitro, por lo que también se denominó "hepatopoyetina" (253). Debido a su efecto mitógeno in vivo e in vitro, es el factor de crecimiento que ha suscitado mayor interés. Además de los efectos mitógenos tiene efectos motógenos, tróficos, antiapoptóticos, angiogénicos y morfogénicos en el desarrollo del hígado, cerebro, placenta, pulmones, intestino, miocardio y sistema reproductivo (211,253,254).
El HGF se sintetiza por las células mesenquimales y se almacena como forma precursora pro-HGF en la matriz extracelular (ME). El HGF es una glicoproteína muy similar a los factores de coagulación y de la fibrinolisis (plasminógeno). La activación del pro-HGF se debe al efecto proteolítico del activador del plasminógeno tipo uroquinasa (μ-PA) (2,4,69,255-261).
Tras una lesión hepática aguda se produce un aumento brusco -10 a 20 veces- del HGF plasmático. Dicha elevación se debe a la liberación del HGF almacenado en la ME y a la síntesis de HGF por los macrófagos estimulados por la IL-6 y TNF-α. Además, el HGF se sintetiza por células mesenquimales de otros órganos -pulmón, riñón, bazo- lo que confirma su efecto endocrino (2,4,259,260,262-265). El HGF señaliza a través de un receptor tirosina quinasa -C-met- cuya fosforilación se observa entre 1 y 15 minutos tras la hepatectomía, hasta los 60 min. En humanos, se ha descrito una elevación más prolongada del HGF; hasta dos semanas post-hepatectomía. Los niveles más elevados se han descrito en la hepatitis fulminante, hallazgo que cuestionó su posible eficacia terapéutica en dicha situación. Este fenómeno paradójico -incapacidad para estimular la proliferación de hepatocitos y prevenir su muerte celular, con niveles muy elevados de HGF- se ha atribuido al "bloqueo del receptor C-met" ante el aumento simultáneo -inhibición competitiva- de otras moléculas de señalización como IL-6 y TGF-β1 en el plasma, o bien al fenómeno ya descrito como "respuesta el estrés hiperproliferativo", por el que un estímulo mitogénico excesivo provoca apoptosis celular mediante la activación del p53 (61,266-270).
La infusión sistémica e intraportal de HGF en roedores, aumenta la síntesis de DNA en los hepatocitos de la zona 1, y la infusión intraportal de HGF "humano" en ratones, provoca la proliferación de los hepatocitos y hepatomegalia (2,4).
La transfección del gen humano -HGF- mediante inyección hidrodinámica del plásmido DNA en el ratón induce hepatomegalia por la activación de la vía β-catenina; y la infusión de cantidades elevadas de HGF aumenta el tamaño del hígado, asociado con el estímulo de las mitogénesis. La retirada de HGF generó una apoptosis marcada y una reinstauración del DNA hepático a los niveles basales (2,4,255,271).
El "pretratamiento" con colagenasa en ratas que posteriormente recibieron HGF, potenció el efecto de HGF. Asimismo, se observó que los hepatocitos aislados mediante la digestión del parénquima con colagenasa, expresaban precozmente marcadores de inicio del ciclo celular. Dicho efecto "preproliferativo" es compatible con el fenómeno de adquisición de competencia -fase muy inicial de G1- descrito por Pardee en 1989, por el que los hepatocitos "competentes" responden más rápidamente a un estímulo regenerativo posterior (1,4,73).
Se ha sugerido que la remodelación de la matriz extracelular -liberación de metaloproteinasas- sería una fase inicial de la regeneración, sensibilizando los hepatocitos al HGF almacenado en el hígado. Probablemente la remodelación de la matriz extracelular, mediada por uroquinasa y metalo-proteinasas, generan la liberación de DAMP -derivados del daño tisular-, capaces de generar una respuesta inflamatoria estéril mediante la activación de los receptores tipo Toll y consecuentemente la activación de los factores de transcripción NF-Kβ, AP-1 y STAT-3 relacionados con el inicio del ciclo celular (54,55,59).
El receptor de membrana con actividad tirosina-quinasa "C-met" está presente en las células epiteliales y mesenquimales. La activación corriente abajo del receptor C-met promueve la cascada de las quinasas activadas por mitógenos (MAP quinasas) y esta estimula los factores de transcripción citosólicos (AP-1) relacionados con la proliferación y la supervivencia celular. Además, el HGF señaliza a través de la quinasa tipo Janus que activa los factores de transcripción STAT-3, el factor nuclear NF-Kβ y la b-catenina. La unión del HGF con el receptor C-met fosforila la β-catenina, facilita su traslocación al núcleo y la expresión de ciclina D necesaria para la transición G0-G1 del ciclo celular (2,4,188,264,267,268).
En modelos murinos transgénicos se ha confirmado la importancia del HGF y su receptor C-met en la respuesta regenerativa del hígado. Ratones deficientes (null mice) en HGF y en su receptor C-met, fallecen durante la gestación y expresan una disminución del tamaño hepático y de las células parenquimatosas (270). Dos grupos independientes han estudiado la regeneración en ratones KO para C-met en el hígado (KO condicional). En dichos ratones, tras una hepatectomía, se observó una disminución de la síntesis de DNA y una ausencia de señalización de las quinasas activadas por mitógenos (MAP quinasas). En dichos animales la administración de Cl4C generó una alteración en la regeneración y cambios inflamatorios más intensos (197,271).
Factor de crecimiento epidérmico (EGF)
Lo componen una familia de 7 factores entre los que destacan: el factor de crecimiento epidérmico (EGF), la amfiregulina (AR), el factor semejante al EGF ligado a la heparina (HB-EGF) y el factor de crecimiento fibroblástico α (TGF α). Dichos factores se sobreexpresan con una lesión hepática. Señalizan a través de receptores de membrana tirosina-quinasa (RTK): EGFR/ErbB1, HER2/ErbB2, HER3/ErbB3 y HER4/ErbB4, y culminan con la activación corriente debajo de la cascada activada por mitógenos (MAPK quinasa) (4,73,272).
Los ratones carentes de receptores de EGF fallecen entre la mitad de la gestación y los primeros 20 días post-natales con defectos en la placenta, cerebro, piel y pulmón (273). En animales en los que se inhibió los receptores del EGF, tras una hepatectomía, fallecían 1/3 y los que sobrevivían mostraban un retraso en la división de los hepatocitos (274). Sin embargo, finalmente tenía lugar la regeneración completa del hígado, sugiriendo que la señalización del receptor EGF es importante pero no esencial para la RH (61,274).
El resto de los factores de la familia EGF también se sintetizan como pro-factores que permanecen fijados a la membrana de los hepatocitos de la que son liberados mediante una proteolisis. Tras una hepatectomía se elevan los niveles plasmáticos de EGF aumentando el cociente EGF/EGFR, confirmando que el EGFR ejerce un papel mitógeno en la fase inicial de la regeneración (4).
El papel del factor HB-EGF se ha estudiado en modelos KO y en animales transgénicos. En el primer caso se objetivó un retraso en la proliferación de los hepatocitos y, en el segundo, se estimulaba la proliferación y se aceleraba la regeneración. Animales carentes de amfiregulina mostraron una alteración de la respuesta regenerativa tras una hepatectomía (275,276).
El TFG-α se sintetiza por los propios hepatocitos -efecto autocrino- y tiene efectos paracrinos sobre las células endoteliales y del epitelio biliar. Tras una hepatectomía, los niveles de TFG-α aumentan entre las 24 y 48 horas. Se ha observado que in vitro e in vivo estimula la síntesis de DNA y, que la adición de TFG-α en cultivos de hepatocitos, estimula la transición a través del punto de control "R" (277,278).
Sin embargo, ratones homozigotos con una deleción en el gen TFG-α expresan una regeneración normal, indicando que el TFG-α es "dispensable" en la regeneración, reforzando su carácter "robusto". En un modelo murino, con sobre-expresión de TFG-α humano, se objetivó un aumento de la proliferación y hepatomegalia (279,280,281).
Cese de la regeneración y regulación del tamaño hepático
La regeneración termina con la restauración de la masa hepática inical necesaria para realizar las funciones hepáticas (índice hepático ~ 2,5 %). La regulación estricta de dicho índice es una de las propiedades más sorprendentes ya que representa un control estricto del cese del ciclo celular y una remodelación del tejido "neoformado" (2,4,5,42,61,72,73).
El cese de la regeneración se ha relacionado con citocinas antiinflamatorias, proapoptóticas y con factores "hepatostáticos" como la IL-10, las proteínas supresoras de señalización de citocinas (SOCS-3), el inhibidor del activador del plasminógeno PAI-1 y especialmente el TGF-β (5,6,61,282).
Estudios con microarrays en roedores y en el cerdo, han confirmado que a la sobreexpresión inicial de genes pro-mitóticos le sucede una suplantación por genes metabólicos y posteriormente por los relacionados con el cese de la proliferación y el retorno al estadio previo quiescente (34,41,61,72,73).
El TGF-β comprende una familia de citocinas (TGF-β1-3) relacionadas con el desarrollo y la cicatrización. Señalizan a través de dos receptores (tipos I y II) que activan a los reguladores de la transcripción, conocidos como proteínas SMAD (Small Mothers Against Decapentaplegic), que se translocan al núcleo. En la mayoría de los tejidos, el TGF-β inhibe la proliferación en la fase G1 del ciclo (283-285).
Se ha descrito que tras una hepatectomía, el ARNm de TGF-β aumenta precozmente, aunque se produce una resistencia transitoria de los hepatocitos al TGF-β por una disminución en la expresión de sus receptores y una sobreexpresión de sus inhibidores. Dicha resistencia remite tras la síntesis de DNA, recuperando la sensibilidad al TGF-β que inhibe la proliferación celular e induce el cese de la regeneración (en la rata a los 5-7 días, y tres semanas en el cerdo) (286-288).
En modelos trangénicos y en animales a los que se infundía directamente TGF-β en diferentes fases de la regeneración, se han confirmado sus efectos antiproliferativos, aunque debido al carácter pleiotrópico de la regeneración, no se observaron cambios relevantes. Ratones transgénicos con sobreexpresión de los receptores de TGF-β, sólo sufrían un retraso en la proliferación celular (61,289,290).
Más significativo es que ratones KO para el receptor II de TGF-β (TGFR2 -/-) muestran una síntesis precoz e incrementada de DNA -primeras 120 horas tras hepatectomía- pero tampoco expresaban diferencias en el cese de la regeneración respecto a los animales control, quizá por la existencia de otras vías inhibitorias de la síntesis de DNA (291).
Otros miembros de la superfamilia TGF-β son las activinas, siendo la más frecuente la activina A. Se identificó en 1993 como un factor proapoptótico de los hepatocitos in vitro. La activina A induce la activación intracelular de SMAD de forma similar a TGF-β, por lo que se considera que ejercen funciones complementarias. La inyección de follistatina, un antagonista del receptor de la activina A tras una hepatectomía, provocó un aumento de la proliferación hepatocitaria y del peso hepático, confirmando que la activina A inhibe la proliferación y un posible regulador del final de la regeneración (5,61,73,292).
En hepatocitos en cultivo, y en modelos "bioartificiales", se ha resaltado el papel de la matriz extracelular en la diferenciación y proliferación de los hepatocitos (60,61,73). A pesar de ser el 0,5 % del peso hepático, la ME tiene un papel activo en la respuesta al daño tisular y en la regeneración. La ME, además de albergar factores de crecimiento -PDGF, TGF-β, VEGF, HGF-, contiene citocinas, metaloproteinasas -colagenasa, gelatinasa- y macromoléculas como fibronectina, laminina y colágena (tipos I, III y VI), muy reactivas cuando se altera la barrera endotelio-sinusoidal (184,185,293-298). Tras una lesión hepática -trauma quirúrgico, isquemia, tóxico, etc.- se libera el activador del plasminógeno tipo uroquinasa (μ-PA) y las metaloproteinasas, que liberan los factores de crecimiento de sus formas precursoras (pro-HGF → HGF) y degradan macromoléculas (fibrinógeno, fibronectina, laminina) con la liberación de moléculas -DAMP- que inducen la respuesta inflamatoria estéril (teoría DANGER) e iniciando la fase de purgado de los hepatocitos (60,156,157,185).
Se ha descrito que la proliferación inicial tras una hepatectomía se asocia a una degradación de la matriz extracelular; dando lugar a acúmulos de hepatocitos neoformados, con un acceso limitado a los factores endocrinos y paracrinos que, en condiciones basales, inhiben la división celular. Los acúmulos hipertróficos requieren una remodelación tisular posterior hasta alcanzar el índice hepático inicial (72).
El grupo de Michalopoulos ha confirmado los efectos in vitro de una proteína inhibidora de la proliferación de los hepatocitos (integrina ligada a una quinasa, ILK) y han descrito que animales KO hepato-específicos para dicha proteína desarrollan un aumento de la proliferación y hepatomegalia (42,299). La ablación hepato-específica de dicha integrina altera el cese de la regeneración. A los 14 días de una hepatectomía, el hígado de los ratones deficientes en ILK alcanza un peso superior al 58 % del peso inicial. En dichos ratones se objetivó un aumento en la expresión de HGF y su receptor C-met, así como una disminución del inhibidor del ciclo celular p27 (299-302).
Debido al gran interés en el desarrollo de "hígados bioartificiales" mediante la "reconstitución" de las matrices de hígados previamente descelularizados con hepatocitos y/o células pluripotenciales, se ha impulsado su desarrollo ante la demanda creciente de injertos para trasplante tanto en el hígado, como riñón, pulmón y corazón (293-298,303,304).
p53 y control de la regeneración
En los organismos pluricelulares existen mecanismos de control del ciclo celular ante situaciones de estrés -falta de nutrientes, hipoxia, lesión del DNA- que detienen el ciclo o inducen la apoptosis celular (305,306).
El sistema más común es la familia de la proteína p53, conocido como "el guardián del genoma" ya que responde al daño del DNA y a otras situaciones de estrés (307-311) Aunque el p53 se conoce como un factor "oncosupresor" (se han referido más de 25.000 mutaciones de p53 en tumores humanos), recientemente ha suscitado interés por su papel en el control y cese de la RH (312).
Kurina y cols. han descrito que el gen p53 es necesario para revertir el alto grado de ploidía y aneuploidía observadas en situaciones basales y en la RH. La ausencia de p53 (ratones p53 -/-) se asoció con un grado muy superior de ploidía (8n y 16n) debido a fallos en la fase final del ciclo o en la citocinesis. Dichos autores han descrito, por primera vez, que p53 regula la expresión de los genes involucrados en las tres fases de la división celular, inicio-progresión, división y regreso al estado G0 tras la hepatectomía (117).
Es bien conocido que la activación de p53 detiene el ciclo celular e induce la apoptosis en situaciones de estrés celular, como la hipoxia severa, acidosis o ante un estímulo mitogénico excesivo. Este último mecanismo es conocido como "respuesta al estrés hiperproliferativo" (307,308) (Fig. 7). El aumento de la actividad de p53 incrementa la expresión del inhibidor p21 del ciclo celular y de las proteínas proapoptóticas (caspasas), causando una parada permanente del ciclo celular e incluso la muerte celular (307).

La actividad proliferativa de proteínas mitogénicas como Myc y Ras -"oncoproteínas"- solamente es posible en la ausencia de p53. La respuesta celular al estrés hiperproliferativo es el correlato in vivo del descrito in vitro como senescencia replicativa. Las células en cultivo, tras varias divisiones, sufren la detención estable del ciclo celular -senescencia replicativa- por el incremento de p53. La senescencia se ha atribuido al estrés hiperproliferativo -Myc y Ras hiperactivos- o a las condiciones poco fisiológicas in vitro: ausencia de los componentes de la matriz extracelular o niveles de oxígeno inadecuados. Por el contrario, células carentes de p53 proliferan indefinidamente en cultivos y se les ha denominado "inmortales" (313-317).
Aunque la respuesta al estrés hiperproliferativo se ha relacionado con mecanismos de protección frente al cáncer, dicha respuesta fisiológica podría producirse en situaciones con un estímulo mitogénico excesivo como el "síndrome de hígado pequeño", el fallo hepático fulminante, la insuficiencia hepática post-hepatectomía o la insuficiencia hepática sobre crónica. En dichas situaciones se produce un gran estímulo mitogénico derivado del daño endotelial -"estrés de rozamiento"-, la necrosis, hemorragia parenquimatosa, vasoespasmo arterial e hipoperfusión (150,151,318-320).
Hoy día se desconoce el porqué en unos casos el hígado regenera, y en otros, fracasa, desarrollando una insuficiencia hepática irreversible; y tampoco existe un criterio nítido sobre la cantidad mínima exigida de hígado remanente en una resección hepática o en el trasplante entre vivos (10,151,321-324). A pesar de la multitud de trabajos sobre la regeneración hepática, sus mecanismos de regulación continúan siendo un misterio (2,4-6,9).
Conclusiones y perspectivas futuras
La regeneración hepática es uno de los fenómenos biológicos más enigmáticos y extraordinarios de adaptación y respuesta para mantener la homeostasis interna. En las últimas décadas ha sido motivo de una investigación intensa debido a sus implicaciones terapéuticas. Se trata de un fenómeno muy complejo y estrictamente regulado que obedece al patrón de respuesta al daño tisular, con una fase inicial de preparación o "cebado" que corresponde a la transición G0-G1 del ciclo celular de los hepatocitos, y una fase posterior de proliferación -fases S y M- que termina con el restablecimiento de la masa hepática. Dicho proceso se ha relacionado con hormonas (insulina/glucagon), citocinas (TNF-α, IL-6, IL-1β, IL-10), factores de crecimientos (HGF, EGF, VEGF) y, en la última década, con células madre hematopoyéticas mesenquimales (CD133+, CD) y células ovales.
La generación reciente de estructuras con fenotipo "hepatoide" en roedores a partir de células madre pluripotenciales inducidas (iPS), ha generado grandes expectativas como posible alternativa al trasplante hepático; aunque debido a la extraordinaria complejidad del hígado deben ser vistas con cautela (325,326).
En la RH se cumplen rigurosamente las características de los sistemas biológicos más regulados (robustness) -pleiomorfismo, redundancia y mecanismo de retroalimentación- como el ciclo celular, la respuesta inmune natural o el ritmo circadiano, y que explica la dificultad de su manipulación con fines terapéuticos.
Debido a la complejidad del hígado, los intentos de suplir sus funciones de forma temporal y definitiva han sido fallidos, salvo el trasplante ortotópico de hígado. Quizá el desarrollo de la terapia celular y el conocimiento de las bases moleculares y fisiológicas de la regeneración, podrán conseguir el sueño anhelado de utilizar la capacidad regenerativa del hígado en el tratamiento de hepatopatías, cuya única alternativa actual es el trasplante hepático.
La frase de Santiago Ramón y Cajal: "en general puede afirmarse que no hay cuestiones agotadas, sino hombres agotados en las cuestiones" es muy oportuna en el intento de desvelar el misterio de la regeneración hepática (327).
Agradecimientos
Los autores agradecen a Lydia Munárriz el excelente trabajo en la edición y transcripción del manuscrito; y a Fabiola de Goñi y Beatriz Urbelz por su colaboración en la transcripción bibliográfica. Los autores expresan el reconocimiento a los investigadores del Área de Hepatología del Centro de Investigación Médica Aplicada por sus valiosas sugerencias y aportaciones. Rogamos nos disculpen aquellos colegas cuyos trabajos no hemos citado por limitaciones de espacio.
![]() Correspondence:
Correspondence:
Javier A.-Cienfuegos.
Departamento de Cirugía General.
Clínica Universidad de Navarra.
Avda. Pío XII, n.o 36.
31008 Pamplona
e-mail: fjacien@unav.es
Received: 19-07-2013
Accepted: 12-11-2013
Bibliografía
1. Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology 2006;43:S45-53. [ Links ]
2. Fausto N, Campbell J S. Liver regeneration. En: Arias IM, Alter HJ, Boyer JL, Cohen DE, Fausto N, Shafritz DA, et al, editores. The liver: biology and pathobiology. 5th ed. Oxford: Wiley & Sons; 2009. p. 549-65. [ Links ]
3. Michalopoulos GK. Liver regeneration: alternative epithelial pathways. Int J Biochem Cell Biol 2011;43:173-9. [ Links ]
4. Michalopoulos GK. Principles of liver regeneration and growth homeostasis. Compr Physiol 2013;3:485-513. [ Links ]
5. Nevzorova YA, Trautwein C. Liver regeneration. En: Boyer TD, Wright TL, Manns MP, Zakim D, editores. Zakim and Boyer's hepatology: A textbook of liver disease. 6th ed. Philadelphia: Saunders Elsevier; 2012. p. 20-35. [ Links ]
6. Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol 2004;5:836-47. [ Links ]
7. Karp SJ. Clinical implications of advances in the basic science of liver repair and regeneration. Am J Transplant 2009;9:1973-80. [ Links ]
8. Curado S, Stainier DY. deLiver'in regeneration: Injury response and development. Semin Liver Dis 2010;30:288-95. [ Links ]
9. De Jonge J, Olthoff K M. Liver regeneration: Mechanisms and clinical relevance. In: Jarnagin WR, Belghiti J, Büchler MW, Chapman WC, D'Angelica MI, DeMatteo RP, et al, editors. Blumgart's surgery of the liver, biliary tract, and pancreas. 5th ed. Philadelphia: Elsevier; 2012. p. 87-101. [ Links ]
10. Clavien PA, Petrowsky H, DeOliveira ML, Graf R. Strategies for safer liver surgery and partial liver transplantation. N Engl J Med 2007;356:1545-59. [ Links ]
11. Adam R, Laurent A, Azoulay D, Castaing D, Bismuth H. Two-stage hepatectomy: A planned strategy to treat irresectable liver tumors. Ann Surg 2000;232:777-85. [ Links ]
12. Abdalla EK. Portal vein embolization (prior to major hepatectomy) effects on regeneration, resectability, and outcome. J Surg Oncol 2010;102:960-7. [ Links ]
13. Dutkowski P, De Rougemont O, Mullhaupt B, Clavien PA. Current and future trends in liver transplantation in Europe. Gastroenterology 2010;138:802,9.e1-4. [ Links ]
14. Brown RS, Jr. Live donors in liver transplantation. Gastroenterology 2008;134:1802-13. [ Links ]
15. Vagefi PA, Parekh J, Ascher NL, Roberts JP, Freise CE. Outcomes with split liver transplantation in 106 recipients: The University of California, San Francisco, experience from 1993 to 2010. Arch Surg 2011;146:1052-9. [ Links ]
16. Schnitzbauer AA, Lang SA, Goessmann H, Nadalin S, Baumgart J, Farkas SA, et al. Right portal vein ligation combined with in situ splitting induces rapid left lateral liver lobe hypertrophy enabling 2-staged extended right hepatic resection in small-for-size settings. Ann Surg 2012;255:405-14. [ Links ]
17. Lafuente S, Bertran MJ, Escorsell A. Artificial liver support. Literature review. Med Clin (Barc) 2011;136:484-7. [ Links ]
18. Gupta S. Stem cells and hepatocyte transplantation. En: Boyer TD, Wright TL, Manns MP, Zakim D, editors. Zakim and Boyer's hepatology: a textbook of liver disease. 6th ed. Philadelphia: Saunders Elsevier; 2012. p. 157-70. [ Links ]
19. Thorgeirsson SS, Grisham JW. Hematopoietic cells as hepatocyte stem cells: A critical review of the evidence. Hepatology 2006;43:2-8. [ Links ]
20. Dhawan A, Puppi J, Hughes RD, Mitry RR. Human hepatocyte transplantation: Current experience and future challenges. Nat Rev Gastroenterol Hepatol 2010;7:288-98. [ Links ]
21. Shafritz DA, Oertel M, Dabeva M D, Grompe M. Liver repopulation by cell transplantation and the role of stem cells. En: Arias IM, Alter HJ, Boyer JL, Cohen DE, Fausto N, Shafritz DA, et al, editors. The liver: biology and pathobiology. 5th ed. Oxford: Wiley & Sons; 2009. p. 577-95. [ Links ]
22. Esquilo. Prometeo encadenado. En: Esquilo, Sófocles, Eurípides, editores. Obras completas. Madrid: Cátedra; 2004. p. 85-126. [ Links ]
23. Tiniakos DG, Kandilis A, Geller SA. Tityus: A forgotten myth of liver regeneration. J Hepatol 2010;53:357-61. [ Links ]
24. Tillmanns H. Experimentelle und anatomische Untersuchungen über Wunden der Leber und Niere. Ein Beitrag zur Lehre von der antiseptischen Wundheilung. Arch Pathol Anat Physiol Klin Med 1879;78:437-74. [ Links ]
25. Knolle P. Liver and immune system. En: Boyer TD, Wright TL, Manns MP, Zakim D, editors. Zakim and Boyer's hepatology: A textbook of liver disease. 6th ed. Philadelphia: Saunders Elsevier; 2012. p. 129-41. [ Links ]
26. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol 2009;27:147-63. [ Links ]
27. Gillilland MG, Young V B, Huffnagle G B. Gastrointestinal microbial ecology with perspectives on health and disease. En: Johnson LR, Ghishan FK, Kaunitz JD, Merchant JL, Said HM, Wood JD, editors. Physiology of the gastrointestinal tract. Vol.1. 5th ed. Amsterdam: Elsevier; 2012. p. 1119-34. [ Links ]
28. Gonzalez-Navajas JM, Frances R, Such J. Bacterial DNA in patients with cirrhosis and sterile ascites. Its role as a marker of bacterial translocation and prognostic tool. Rev Esp Enferm Dig 2007;99:599-603. [ Links ]
29. Jia C. Advances in the regulation of liver regeneration. Expert Rev Gastroenterol Hepatol 2011;5:105-21. [ Links ]
30. Riehle KJ, Dan YY, Campbell JS, Fausto N. New concepts in liver regeneration. J Gastroenterol Hepatol 2011;26(Supl. 1):203-12. [ Links ]
31. Pahlavan PS, Feldmann RE,Jr, Zavos C, Kountouras J. Prometheus' challenge: Molecular, cellular and systemic aspects of liver regeneration. J Surg Res 2006;134:238-51. [ Links ]
32. Morgan DO. The cell cycle. En: Morgan DO, editor. The cell cycle: Principles of control. London: New Science Press; 2007. p. 1-9. [ Links ]
33. Satyanarayana A, Geffers R, Manns MP, Buer J, Rudolph KL. Gene expression profile at the G1/S transition of liver regeneration after partial hepatectomy in mice. Cell Cycle 2004;3:1405-17. [ Links ]
34. Chauhan A, Lorenzen S, Herzel H, Bernard S. Regulation of mammalian cell cycle progression in the regenerating liver. J Theor Biol 2011;283:103-12. [ Links ]
35. Duncan AW. Aneuploidy, polyploidy and ploidy reversal in the liver. Semin Cell Dev Biol 2013;24:347-56. [ Links ]
36. Higgins GM, Anderson RM. Experimental pathology of the liver. 1. Restoration of the liver of the white rat following partial surgical removal, Arch Pathol 1931;12:186-8. [ Links ]
37. Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc 2008;3:1167-70. [ Links ]
38. Martins PN, Theruvath TP, Neuhaus P. Rodent models of partial hepatectomies. Liver Int 2008;28:3-11. [ Links ]
39. Fausto N. Lessons from genetically engineered animal models. V. Knocking out genes to study liver regeneration: present and future. Am J Physiol 1999;277:G917-21. [ Links ]
40. Mitchell C, Gilgenkrantz H. Transcriptional profiling of liver regeneration: New approaches to an old trick! J Hepatol 2003;38:847-9. [ Links ]
41. Fukuhara Y, Hirasawa A, Li XK, Kawasaki M, Fujino M, Funeshima N, et al. Gene expression profile in the regenerating rat liver after partial hepatectomy. J Hepatol 2003;38:784-92. [ Links ]
42. Kurinna S, Barton MC. Hierarchies of transcriptional regulation during liver regeneration. Prog Mol Biol Transl Sci 2010;97:201-27. [ Links ]
43. Locker J, Tian J, Carver R, Concas D, Cossu C, Ledda-Columbano GM, et al. A common set of immediate-early response genes in liver regeneration and hyperplasia. Hepatology 2003;38:314-25. [ Links ]
44. Fausto N, Campbell JS, Riehle KJ. Liver regeneration. J Hepatol 2012;57:692-4. [ Links ]
45. Call MK, Tsonis PA. Vertebrate limb regeneration. Adv Biochem Eng Biotechnol 2005;93:67-81. [ Links ]
46. Matthews RP. Zebrafish as a model system for the study of liver development and disease. En: Arias IM, Alter HJ, Boyer JL, Cohen DE, Fausto, Shafritz DA, et al, editors. The liver: biology and pathobiology. 5th ed. Oxford: Wiley & Sons; 2009. p. 1067-74. [ Links ]
47. Carlson BM. Some principles of regeneration in mammalian systems. Anat Rec B New Anat 2005;287:4-13. [ Links ]
48. Morrison JI, Loof S, He P, Simon A. Salamander limb regeneration involves the activation of a multipotent skeletal muscle satellite cell population. J Cell Biol 2006;172:433-40. [ Links ]
49. Otu HH, Naxerova K, Ho K, Can H, Nesbitt N, Libermann TA, et al. Restoration of liver mass after injury requires proliferative and not embryonic transcriptional patterns. J Biol Chem 2007;282:11197-204. [ Links ]
50. Mastellos D, Lambris J D. Integrating innate immunity into a global systems context: the complement paradigm shift. En: Rigoutsos I, Stephanopoulus G, editors. Systems Biology. Vol. 2. Networks, models, and applications. Oxford: Oxford University Press; 2007. p. 169-92. [ Links ]
51. Chu J, Sadler KC. New school in liver development: Lessons from zebrafish. Hepatology 2009;50:1656-63. [ Links ]
52. Miyaoka YF, Ebato KF, Kato HF, Arakawa SF, Shimizu SF, Miyajima A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol 2012;22:1166-75. [ Links ]
53. Grisham JW. A morphologic study of deoxyribonucleic acid synthesis and cell proliferation in regenerating rat liver; autoradiography with thymidine-H3. Cancer Res 1962;22:842-9. [ Links ]
54. Rock KL, Lai JJ, Kono H. Innate and adaptive immune responses to cell death. Immunol Rev 2011;243:191-205. [ Links ]
55. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol 2010;28:321-42. [ Links ]
56. Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol 2011;29:185-214. [ Links ]
57. Lemasters JJ. Dying a thousand deaths: Redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology 2005;129:351-60. [ Links ]
58. Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: A tale of two deaths? Hepatology 2006;43:S31-44. [ Links ]
59. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology 2012;143:1158-72. [ Links ]
60. Mohammed FF, Khokha R. Thinking outside the cell: Proteases regulate hepatocyte division. Trends Cell Biol 2005;15:555-63. [ Links ]
61. Bohm F, Kohler UA, Speicher T, Werner S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol Med 2010;2:294-305. [ Links ]
62. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010;140:805-20. [ Links ]
63. Medzhitov R. Inflammation 2010: New adventures of an old flame. Cell 2010;140:771-6. [ Links ]
64. Calfee CS, Matthay MA. Clinical immunology: Culprits with evolutionary ties. Nature 2010;464:41-2. [ Links ]
65. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med 2009;361:1570-83. [ Links ]
66. Mohammed FF, Pennington CJ, Kassiri Z, Rubin JS, Soloway PD, Ruther U, et al. Metalloproteinase inhibitor TIMP-1 affects hepatocyte cell cycle via HGF activation in murine liver regeneration. Hepatology 2005;41:857-67. [ Links ]
67. Christophi C, Harun N, Fifis T. Liver regeneration and tumor stimulation -a review of cytokine and angiogenic factors. J Gastrointest Surg 2008;12:966-80. [ Links ]
68. Malato Y, Naqvi S, Schurmann N, Ng R, Wang B, Zape J, et al. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J Clin Invest 2011;121:4850-60. [ Links ]
69. Morgan DO. The cell-cycle control system. En: Morgan DO, editor. The cell cycle: Principles of control. London: New Science Press; 2007. p. 27-55. [ Links ]
70. Massague J. G1 cell-cycle control and cancer. Nature 2004;432:298-306. [ Links ]
71. Michalopoulos GK. Liver regeneration after partial hepatectomy: Critical analysis of mechanistic dilemmas. Am J Pathol 2010;176:2-13. [ Links ]
72. Kurinna S, Barton MC. Cascades of transcription regulation during liver regeneration. Int J Biochem Cell Biol 2011;43:189-97. [ Links ]
73. Michalopoulos GK, DeFrances M. Liver regeneration. Adv Biochem Eng Biotechnol 2005;93:101-34. [ Links ]
74. Duncan AW, Hickey RD, Paulk NK, Culberson AJ, Olson SB, Finegold MJ, et al. Ploidy reductions in murine fusion-derived hepatocytes. PLoS Genet 2009;5:e1000385. [ Links ]
75. Duncan AW, Hanlon Newell AE, Smith L, Wilson EM, Olson SB, Thayer MJ, et al. Frequent aneuploidy among normal human hepatocytes. Gastroenterology 2012;142:25-8. [ Links ]
76. Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010;468:310-5. [ Links ]
77. Maeno H, Ono T, Dhar DK, Sato T, Yamanoi A, Nagasue N. Expression of hypoxia inducible factor-1alpha during liver regeneration induced by partial hepatectomy in rats. Liver Int 2005;25:1002-9. [ Links ]
78. Shimizu H, Miyazaki M, Wakabayashi Y, Mitsuhashi N, Kato A, Ito H, et al. Vascular endothelial growth factor secreted by replicating hepatocytes induces sinusoidal endothelial cell proliferation during regeneration after partial hepatectomy in rats. J Hepatol 2001;34:683-9. [ Links ]
79. Drixler TA, Vogten MJ, Ritchie ED, van Vroonhoven TJ, Gebbink MF, Voest EE, et al. Liver regeneration is an angiogenesis-associated phenomenon. Ann Surg 2002;236:703,11; discussion 711-2. [ Links ]
80. Drixler TA, Vogten JM, Gebbink MF, Carmeliet P, Voest EE, Borel Rinkes IH. Plasminogen mediates liver regeneration and angiogenesis after experimental partial hepatectomy. Br J Surg 2003;90:1384-90. [ Links ]
81. Vollmar B, Menger MD. The hepatic microcirculation: Mechanistic contributions and therapeutic targets in liver injury and repair. Physiol Rev 2009;89:1269-339. [ Links ]
82. Wu Y, Guo F, Liu J, Xiao X, Huang L, He D. Triple labeling with three thymidine analogs reveals a well-orchestrated regulation of hepatocyte proliferation during liver regeneration. Hepatol Res 2011;41:1230-9. [ Links ]
83. Ferri D, Moro L, Mastrodonato M, Capuano F, Marra E, Liquori GE, et al. Ultrastructural zonal heterogeneity of hepatocytes and mitochondria within the hepatic acinus during liver regeneration after partial hepatectomy. Biol Cell 2005;97:277-88. [ Links ]
84. Petersen BE, Zajac VF, Michalopoulos GK. Hepatic oval cell activation in response to injury following chemically induced periportal or pericentral damage in rats. Hepatology 1998;27:1030-8. [ Links ]
85. Dabeva MD, Shafritz DA. Hepatic stem cells and liver repopulation. Semin Liver Dis 2003;23:349-62. [ Links ]
86. Roskams TA, Libbrecht L, Desmet VJ. Progenitor cells in diseased human liver. Semin Liver Dis 2003;23:385-96. [ Links ]
87. Van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 2009;71:241-60. [ Links ]
88. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663-76. [ Links ]
89. Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc 2007;2:3081-9. [ Links ]
90. Cherry AB, Daley GQ. Reprogrammed cells for disease modeling and regenerative medicine. Annu Rev Med 2013;64:277-90. [ Links ]
91. Bhave VS, Paranjpe S, Bowen WC, Donthamsetty S, Bell AW, Khillan JS, et al. Genes inducing iPS phenotype play a role in hepatocyte survival and proliferation in vitro and liver regeneration in vivo. Hepatology 2011;54:1360-70. [ Links ]
92. Liu T, Wang Y, Tai G, Zhang S. Could co-transplantation of iPS cells derived hepatocytes and MSCs cure end-stage liver disease? Cell Biol Int 2009;33:1180-3. [ Links ]
93. Locke JE, Shamblott MJ, Cameron AM. Stem cells and the liver: Clinical applications in transplantation. Adv Surg 2009;43:35-51. [ Links ]
94. Sancho-Bru P, Najimi M, Caruso M, Pauwelyn K, Cantz T, Forbes S, et al. Stem and progenitor cells for liver repopulation: Can we standardise the process from bench to bedside? Gut 2009;58:594-603. [ Links ]
95. Asgari S, Pournasr B, Salekdeh GH, Ghodsizadeh A, Ott M, Baharvand H. Induced pluripotent stem cells: A new era for hepatology. J Hepatol 2010;53:738-51. [ Links ]
96. Diehl AM, Chute J. Underlying potential: cellular and molecular determinants of adult liver repair. J Clin Invest 2013;123:1858-60. [ Links ]
97. DeLeve LD. Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest 2013;123:1861-6. [ Links ]
98. Yu Y, Fisher JE, Lillegard JB, Rodysill B, Amiot B, Nyberg SL. Cell therapies for liver diseases. Liver Transpl 2012;18:9-21. [ Links ]
99. Furst G, Schulte am Esch J, Poll LW, Hosch SB, Fritz LB, Klein M, et al. Portal vein embolization and autologous CD133+ bone marrow stem cells for liver regeneration: Initial experience. Radiology 2007;243:171-9. [ Links ]
100. Mertsching H, Schanz J, Steger V, Schandar M, Schenk M, Hansmann J, et al. Generation and transplantation of an autologous vascularized bioartificial human tissue. Transplantation 2009;88:203-10. [ Links ]
101. Orlando G, Wood KJ, Stratta RJ, Yoo JJ, Atala A, Soker S. Regenerative medicine and organ transplantation: Past, present, and future. Transplantation 2011;91:1310-7. [ Links ]
102. Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science 2008;322:1490-4. [ Links ]
103. Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, et al. Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat Biotechnol 2007;25:903-10. [ Links ]
104. Overturf K, Al-Dhalimy M, Finegold M, Grompe M. The repopulation potential of hepatocyte populations differing in size and prior mitotic expansion. Am J Pathol 1999;155:2135-43. [ Links ]
105. Starzl TE, Fung J, Tzakis A, Todo S, Demetris AJ, Marino IR, et al. Baboon-to-human liver transplantation. Lancet 1993;341:65-71. [ Links ]
106. Van Thiel DH, Gavaler JS, Kam I, Francavilla A, Polimeno L, Schade RR, et al. Rapid growth of an intact human liver transplanted into a recipient larger than the donor. Gastroenterology 1987;93:1414-9. [ Links ]
107. Vauthey JN, Abdalla EK, Doherty DA, Gertsch P, Fenstermacher MJ, Loyer EM, et al. Body surface area and body weight predict total liver volume in Western adults. Liver Transpl 2002;8:233-40. [ Links ]
108. Morgan DO. Control of cell proliferation and growth. In: Morgan DO, editor. The cell cycle: Principles of control. London: New Science Press; 2007. p. 195-225. [ Links ]
109. Conlon I, Raff M. Size control in animal development. Cell 1999;96:235-44. [ Links ]
110. Jorgensen P, Tyers M. How cells coordinate growth and division. Curr Biol 2004;14:R1014-27. [ Links ]
111. Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010;467:707-10. [ Links ]
112. Gupta S. Hepatic polyploidy and liver growth control. Semin Cancer Biol 2000;10:161-71. [ Links ]
113. Lacroix B, Maddox AS. Cytokinesis, ploidy and aneuploidy. J Pathol 2012;226:338-51. [ Links ]
114. Mitchison JM. Growth during the cell cycle. Int Rev Cytol 2003;226:165-258. [ Links ]
115. Nurse P. Genetic control of cell size at cell division in yeast. Nature 1975;256:547-51. [ Links ]
116. Gentric G, Celton-Morizur S, Desdouets C. Polyploidy and liver proliferation. Clin Res Hepatol Gastroenterol 2012;36:29-34. [ Links ]
117. Kurinna S, Stratton SA, Coban Z, Schumacher JM, Grompe M, Duncan AW, et al. P53 Regulates a mitotic transcription program and determines ploidy in normal mouse liver. Hepatology 2013;57:2004-13. [ Links ]
118. Minamishima YA, Nakayama K, Nakayama K. Recovery of liver mass without proliferation of hepatocytes after partial hepatectomy in Skp2-deficient mice. Cancer Res 2002;62:995-9. [ Links ]
119. Wirth KG, Wutz G, Kudo NR, Desdouets C, Zetterberg A, Taghybeeglu S, et al. Separase: a universal trigger for sister chromatid disjunction but not chromosome cycle progression. J Cell Biol 2006;172:847-60. [ Links ]
120. Edgar BA, Orr-Weaver TL. Endoreplication cell cycles: More for less. Cell 2001;105:297-306. [ Links ]
121. Lu P, Prost S, Caldwell H, Tugwood JD, Betton GR, Harrison DJ. Microarray analysis of gene expression of mouse hepatocytes of different ploidy. Mamm Genome 2007;18:617-26. [ Links ]
122. Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009;460:278-82. [ Links ]
123. Kamel IR, Erbay N, Warmbrand G, Kruskal JB, Pomfret EA, Raptopoulos V. Liver regeneration after living adult right lobe transplantation. Abdom Imaging 2003;28:53-7. [ Links ]
124. Jin MB, Shimamura T, Taniguchi M, Nagasako Y, Suzuki T, Kamiyama T, et al. Liver regeneration in living-donor liver transplantation. Nihon Geka Gakkai Zasshi 2004;105:674-9. [ Links ]
125. Humar A, Kosari K, Sielaff TD, Glessing B, Gomes M, Dietz C, et al. Liver regeneration after adult living donor and deceased donor split-liver transplants. Liver Transpl 2004;10:374-8. [ Links ]
126. Mortensen KE, Conley LN, Hedegaard J, Kalstad T, Sorensen P, Bendixen C, et al. Regenerative response in the pig liver remnant varies with the degree of resection and rise in portal pressure. Am J Physiol Gastrointest Liver Physiol 2008;294:G819-30. [ Links ]
127. Bucher NL, Swaffield MN. The rate of incorporation of labeled thymidine into the deoxyribonucleic acid of regenerating rat liver in relation to the amount of liver excised. Cancer Res 1964;24:1611-25. [ Links ]
128. Weinbren K, Tarsh E. The mitotic response in the rat liver after different regenerative stimuli. Br J Exp Pathol 1964;45:475-80. [ Links ]
129. Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol 2012;30:313-35. [ Links ]
130. Lin E, Calvano SE, Lowry SF. Inflammatory cytokines and cell response in surgery. Surgery 2000;127:117-26. [ Links ]
131. Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med 2003;198:913-23. [ Links ]
132. DeAngelis RA, Markiewski MM, Lambris JD. Liver regeneration: A link to inflammation through complement. Adv Exp Med Biol 2006;586:17-34. [ Links ]
133. Whitacre JM. Biological robustness: Paradigms, mechanisms, and systems principles. Front Genet 2012;3:67. [ Links ]
134. Stelling J, Sauer U, Szallasi Z, Doyle FJ, 3rd, Doyle J. Robustness of cellular functions. Cell 2004;118:675-85. [ Links ]
135. Kitano H. Cancer as a robust system: Implications for anticancer therapy. Nat Rev Cancer 2004;4:227-35. [ Links ]
136. Corlu A, Loyer P. Regulation of the g1/s transition in hepatocytes: Involvement of the cyclin-dependent kinase cdk1 in the DNA replication. Int J Hepatol 2012;2012:689324. [ Links ]
137. Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: Dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol 2003;15:221-31. [ Links ]
138. Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 1997;13:261-91. [ Links ]
139. Albrecht JH, Mullany L K. Cell cycle control in the liver. En: Arias IM, Alter HJ, Boyer JL, Cohen DE, Fausto N, Shafritz DA, et al, editors. The liver: Biology and pathobiology. 5th ed. Oxford: Wiley & Sons; 2009. p. 1015-27. [ Links ]
140. Pardee AB. G1 events and regulation of cell proliferation. Science 1989;246:603-8. [ Links ]
141. Nurse P. Wee beasties. Nature 2004;432:557. [ Links ]
142. Murray AW. Recycling the cell cycle: Cyclins revisited. Cell 2004;116:221-34. [ Links ]
143. Stefater JA,3rd, Ren S, Lang RA, Duffield JS. Metchnikoff's policemen: Macrophages in development, homeostasis and regeneration. Trends Mol Med 2011;17:743-52. [ Links ]
144. Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology 2008;134:1641-54. [ Links ]
145. Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: A clear and present danger. Physiol Rev 2010;90:1165-94. [ Links ]
146. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009;16:3-11. [ Links ]
147. Guicciardi ME, Gores GJ. Apoptosis as a mechanism for liver disease progression. Semin Liver Dis 2010;30:402-10. [ Links ]
148. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 2008;8:279-89. [ Links ]
149. Strey CW, Siegmund B, Rosenblum S, Marquez-Pinilla RM, Oppermann E, Huber-Lang M, et al. Complement and neutrophil function changes after liver resection in humans. World J Surg 2009;33:2635-43. [ Links ]
150. Solis-Munoz P. Acute on chronic liver failure and prognostic factors: Time for reevaluation. Rev Esp Enferm Dig 2011;103:169-76. [ Links ]
151. Helling TS. Liver failure following partial hepatectomy. HPB (Oxford) 2006;8:165-74. [ Links ]
152. Hammond JS, Guha IN, Beckingham IJ, Lobo DN. Prediction, prevention and management of postresection liver failure. Br J Surg 2011;98:1188-200. [ Links ]
153. Antoniades CG, Berry PA, Wendon JA, Vergani D. The importance of immune dysfunction in determining outcome in acute liver failure. J Hepatol 2008;49:845-61. [ Links ]
154. Wasmuth HE, Kunz D, Yagmur E, Timmer-Stranghoner A, Vidacek D, Siewert E, et al. Patients with acute on chronic liver failure display "sepsis-like" immune paralysis. J Hepatol 2005;42:195-201. [ Links ]
155. Van Golen RF, Van Gulik TM, Heger M. The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev 2012;23:69-84. [ Links ]
156. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994;12:991-1045. [ Links ]
157. Matzinger P. An innate sense of danger. Semin Immunol 1998;10:399-415. [ Links ]
158. Matzinger P. The danger model: A renewed sense of self. Science 2002;296:301-5. [ Links ]
159. Mollen KP, Anand RJ, Tsung A, Prince JM, Levy RM, Billiar TR. Emerging paradigm: Toll-like receptor 4-sentinel for the detection of tissue damage. Shock 2006;26:430-7. [ Links ]
160. Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology 2006;130:1886-900. [ Links ]
161. Bianchi ME. DAMPs, PAMPs and alarmins: All we need to know about danger. J Leukoc Biol 2007;81:1-5. [ Links ]
162. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 2004;4:499-511. [ Links ]
163. Zimmermann HW, Trautwein C, Tacke F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol 2012;3:56. [ Links ]
164. Lamkanfi M, Dixit VM. Inflammasomes: Guardians of cytosolic sanctity. Immunol Rev 2009;227:95-105. [ Links ]
165. Leemans JC, Cassel SL, Sutterwala FS. Sensing damage by the NLRP3 inflammasome. Immunol Rev 2011;243:152-62. [ Links ]
166. Trauner M, Halilbasic E. Nuclear receptors as new perspective for the management of liver diseases. Gastroenterology 2011;140:1120-1125.e1-12. [ Links ]
167. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science 2012;335:936-41. [ Links ]
168. Basta G, Navarra T, De Simone P, Del Turco S, Gastaldelli A, Filipponi F. What is the role of the receptor for advanced glycation end products-ligand axis in liver injury? Liver Transpl 2011;17:633-40. [ Links ]
169. Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep 2004;5:825-30. [ Links ]
170. DeAngelis RA, Markiewski MM, Kourtzelis I, Rafail S, Syriga M, Sandor A, et al. A complement-IL-4 regulatory circuit controls liver regeneration. J Immunol 2012;188:641-8. [ Links ]
171. Kohl J. The role of complement in danger sensing and transmission. Immunol Res 2006;34:157-76. [ Links ]
172. Markiewski MM, DeAngelis RA, Strey CW, Foukas PG, Gerard C, Gerard N, et al. The regulation of liver cell survival by complement. J Immunol 2009;182:5412-8. [ Links ]
173. Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol 2007;171:715-27. [ Links ]
174. Leslie M. Immunology. The new view of complement. Science 2012;337:1034-7. [ Links ]
175. Matzinger P, Kamala T. Tissue-based class control: The other side of tolerance. Nat Rev Immunol 2011;11:221-30. [ Links ]
176. Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol 2010;10:131-44. [ Links ]
177. Seki E, Tsutsui H, Iimuro Y, Naka T, Son G, Akira S, et al. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology 2005;41:443-50. [ Links ]
178. Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalk between the liver and gut. J Physiol 2012;590:447-58. [ Links ]
179. Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology 2008;48:322-35. [ Links ]
180. Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006;312:233-6. [ Links ]
181. Cornell RP, Liljequist BL, Bartizal KF. Depressed liver regeneration after partial hepatectomy of germ-free, athymic and lipopolysaccharide-resistant mice. Hepatology 1990;11:916-22. [ Links ]
182. Cornell RP. Restriction of gut-derived endotoxin impairs DNA synthesis for liver regeneration. Am J Physiol 1985;249:R563-9. [ Links ]
183. Cornell RP. Gut-derived endotoxin elicits hepatotrophic factor secretion for liver regeneration. Am J Physiol 1985;249:R551-62. [ Links ]
184. Kim TH, Mars WM, Stolz DB, Petersen BE, Michalopoulos GK. Extracellular matrix remodeling at the early stages of liver regeneration in the rat. Hepatology 1997;26:896-904. [ Links ]
185. Arthur MJ. Matrix degradation in liver: A role in injury and repair. Hepatology 1997;26:1069-71. [ Links ]
186. Diehl AM. Cytokine regulation of liver injury and repair. Immunol Rev 2000;174:160-71. [ Links ]
187. Van Sweringen HL, Sakai N, Tevar AD, Burns JM, Edwards MJ, Lentsch AB. CXC chemokine signaling in the liver: Impact on repair and regeneration. Hepatology 2011;54:1445-53. [ Links ]
188. Hancock JT. Extracellular signals: hormones, cytokines and growth factors. En: Hancock JT, editor. Cell signalling. 3rd ed. Oxford: Oxford University Press; 2010. p. 55-77. [ Links ]
189. Morello D, Fitzgerald MJ, Babinet C, Fausto N. c-myc, c-fos, and c-jun regulation in the regenerating livers of normal and H-2K/c-myc transgenic mice. Mol Cell Biol 1990;10:3185-93. [ Links ]
190. Steer CJ. Liver regeneration. FASEB J 1995;9:1396-400. [ Links ]
191. Li JW, Wang GP, Fan JY, Chang CF, Xu CS. Eight paths of ERK1/2 signalling pathway regulating hepatocyte proliferation in rat liver regeneration. J Genet 2011;90:435-42. [ Links ]
192. Terui K, Ozaki M. The role of STAT3 in liver regeneration. Drugs Today (Barc) 2005;41:461-9. [ Links ]
193. Selzner N, Selzner M, Odermatt B, Tian Y, Van Rooijen N, Clavien PA. ICAM-1 triggers liver regeneration through leukocyte recruitment and Kupffer cell-dependent release of TNF-alpha/IL-6 in mice. Gastroenterology 2003;124:692-700. [ Links ]
194. Levy DE, Lee CK. What does Stat3 do? J Clin Invest 2002;109:1143-8. [ Links ]
195. Li W, Liang X, Kellendonk C, Poli V, Taub R. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem 2002;277:28411-7. [ Links ]
196. Malato Y, Sander LE, Liedtke C, Al-Masaoudi M, Tacke F, Trautwein C, et al. Hepatocyte-specific inhibitor-of-kappaB-kinase deletion triggers the innate immune response and promotes earlier cell proliferation during liver regeneration. Hepatology 2008;47:2036-50. [ Links ]
197. Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci U S A 2004;101:10608-13. [ Links ]
198. Taub R, Greenbaum LE, Peng Y. Transcriptional regulatory signals define cytokine-dependent and -independent pathways in liver regeneration. Semin Liver Dis 1999;19:117-27. [ Links ]
199. Mastellos D, Papadimitriou JC, Franchini S, Tsonis PA, Lambris JD. A novel role of complement: Mice deficient in the fifth component of complement (C5) exhibit impaired liver regeneration. J Immunol 2001;166:2479-86. [ Links ]
200. Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006;107:637-41. [ Links ]
201. Semple JW, Italiano JE,Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol 2011;11:264-74. [ Links ]
202. Nowatari T, Fukunaga K, Ohkohchi N. Regulation of signal transduction and role of platelets in liver regeneration. Int J Hepatol 2012;2012:542479. [ Links ]
203. Nurden AT. Platelets and tissue remodeling: Extending the role of the blood clotting system. Endocrinology 2007;148:3053-5. [ Links ]
204. Tomiyama K, Miyazaki M, Nukui M, Takaishi M, Nakao A, Shimizu N, et al. Limited contribution of cells of intact extrahepatic tissue origin to hepatocyte regeneration in transplanted rat liver. Transplantation 2007;83:624-30. [ Links ]
205. Alkozai EM, Nijsten MW, de Jong KP, de Boer MT, Peeters PM, Slooff MJ, et al. Immediate postoperative low platelet count is associated with delayed liver function recovery after partial liver resection. Ann Surg 2010;251:300-6. [ Links ]
206. Lisman T, Porte RJ. The role of platelets in liver inflammation and regeneration. Semin Thromb Hemost 2010;36:170-4. [ Links ]
207. Lesurtel M, Graf R, Aleil B, Walther DJ, Tian Y, Jochum W, et al. Platelet-derived serotonin mediates liver regeneration. Science 2006;312:104-7. [ Links ]
208. Tian Y, Graf R, El-Badry AM, Lesurtel M, Furrer K, Moritz W, et al. Activation of serotonin receptor-2B rescues small-for-size liver graft failure in mice. Hepatology 2011;53:253-62. [ Links ]
209. Matondo RB, Punt C, Homberg J, Toussaint MJ, Kisjes R, Korporaal SJ, et al. Deletion of the serotonin transporter in rats disturbs serotonin homeostasis without impairing liver regeneration. Am J Physiol Gastrointest Liver Physiol 2009;296:G963-8. [ Links ]
210. Moolten FL, Bucher NL. Regeneration of rat liver: Transfer of humoral agent by cross circulation. Science 1967;158:272-4. [ Links ]
211. Michalopoulos G, Cianciulli HD, Novotny AR, Kligerman AD, Strom SC, Jirtle RL. Liver regeneration studies with rat hepatocytes in primary culture. Cancer Res 1982;42:4673-82. [ Links ]
212. Jirtle RL, Michalopoulos G. Effects of partial hepatectomy on transplanted hepatocytes. Cancer Res 1982;42:3000-4. [ Links ]
213. Ozaki K, Leonard WJ. Cytokine and cytokine receptor pleiotropy and redundancy. J Biol Chem 2002;277:29355-8. [ Links ]
214. Glotzbach JP, Wong VW, Gurtner GC, Longaker MT. Regenerative medicine. Curr Probl Surg 2011;48:148-212. [ Links ]
215. Pennica D, King KL, Shaw KJ, Luis E, Rullamas J, Luoh SM, et al. Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc Natl Acad Sci U S A 1995;92:1142-6. [ Links ]
216. Bustos M, Beraza N, Lasarte JJ, Baixeras E, Alzuguren P, Bordet T, et al. Protection against liver damage by cardiotrophin-1: A hepatocyte survival factor up-regulated in the regenerating liver in rats. Gastroenterology 2003;125:192-201. [ Links ]
217. Bode JG, Heinrich P C. Interleukin-6 signaling during the acute-phase response of the liver. En: Arias IM, Boyer JL, Chisari FV, Fausto N, Schachter D, Shafritz DA, editors. The liver biology and pathobiology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2001. p. 565-80. [ Links ]
218. Szabo G, Romics L, Jr, Frendl G. Liver in sepsis and systemic inflammatory response syndrome. Clin Liver Dis 2002;6:1045,66, x. [ Links ]
219. Streetz KL, Wustefeld T, Klein C, Manns MP, Trautwein C. Mediators of inflammation and acute phase response in the liver. Cell Mol Biol (Noisy-le-grand) 2001;47:661-73. [ Links ]
220. Trautwein C, Boker K, Manns MP. Hepatocyte and immune system: Acute phase reaction as a contribution to early defence mechanisms. Gut 1994;35:1163-6. [ Links ]
221. Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. J Pathol 2008;214:161-78. [ Links ]
222. Kishimoto T, Taga T, Akira S. Cytokine signal transduction. Cell 1994;76:253-62. [ Links ]
223. Lutticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A, et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 1994;263:89-92. [ Links ]
224. Matsuda T, Hirano T. Association of p72 tyrosine kinase with Stat factors and its activation by interleukin-3, interleukin-6, and granulocyte colony-stimulating factor. Blood 1994;83:3457-61. [ Links ]
225. Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996;274:1379-83. [ Links ]
226. Jin X, Zhang Z, Beer-Stolz D, Zimmers TA, Koniaris LG. Interleukin-6 inhibits oxidative injury and necrosis after extreme liver resection. Hepatology 2007;46:802-12. [ Links ]
227. Iniguez M, Berasain C, Martinez-Anso E, Bustos M, Fortes P, Pennica D, et al. Cardiotrophin-1 defends the liver against ischemia-reperfusion injury and mediates the protective effect of ischemic preconditioning. J Exp Med 2006;203:2809-15. [ Links ]
228. Oyama T, Sadamori H, Matsukawa H, Murata H, Umeda Y, Watanabe Y, et al. Small liver graft regenerates through immediate increase of HGF and IL-6--possible involvement of sinusoidal tensile/shear stress in small liver graft. Hepatogastroenterology 2007;54:2078-83. [ Links ]
229. Blindenbacher A, Wang X, Langer I, Savino R, Terracciano L, Heim MH. Interleukin 6 is important for survival after partial hepatectomy in mice. Hepatology 2003;38:674-82. [ Links ]
230. Beraza N, Marques JM, Martinez-Anso E, Iniguez M, Prieto J, Bustos M. Interplay among cardiotrophin-1, prostaglandins, and vascular endothelial growth factor in rat liver regeneration. Hepatology 2005;41:460-9. [ Links ]
231. Tunon MJ, San Miguel B, Crespo I, Riezu-Boj JI, Larrea E, Alvarez M, et al. Cardiotrophin-1 promotes a high survival rate in rabbits with lethal fulminant hepatitis of viral origin. J Virol 2011;85:13124-32. [ Links ]
232. Marques JM, Belza I, Holtmann B, Pennica D, Prieto J, Bustos M. Cardiotrophin-1 is an essential factor in the natural defense of the liver against apoptosis. Hepatology 2007;45:639-48. [ Links ]
233. White P, Brestelli JE, Kaestner KH, Greenbaum LE. Identification of transcriptional networks during liver regeneration. J Biol Chem 2005;280:3715-22. [ Links ]
234. Rodrigues CMP, Castro RE, Steer CJ. The role of bile acids in the modulation of apoptosis. Principles of Medical Biology 2004;15:119-45. [ Links ]
235. Hulzebos CV, Renfurm L, Bandsma RH, Verkade HJ, Boer T, Boverhof R, et al. Measurement of parameters of cholic acid kinetics in plasma using a microscale stable isotope dilution technique: Application to rodents and humans. J Lipid Res 2001;42:1923-9. [ Links ]
236. Palmeira CM, Rolo AP. Mitochondrially-mediated toxicity of bile acids. Toxicology 2004;203:1-15. [ Links ]
237. Monte MJ, Martinez-Diez MC, El-Mir MY, Mendoza ME, Bravo P, Bachs O, et al. Changes in the pool of bile acids in hepatocyte nuclei during rat liver regeneration. J Hepatol 2002;36:534-42. [ Links ]
238. Gravante G, Knowles T, Ong SL, Al-Taan O, Metcalfe M, Dennison AR, et al. Bile changes after liver surgery: Experimental and clinical lessons for future applications. Dig Surg 2010;27:450-60. [ Links ]
239. Chiang JY. Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. J Hepatol 2004;40:539-51. [ Links ]
240. Martinez JD, Stratagoules ED, LaRue JM, Powell AA, Gause PR, Craven MT, et al. Different bile acids exhibit distinct biological effects: The tumor promoter deoxycholic acid induces apoptosis and the chemopreventive agent ursodeoxycholic acid inhibits cell proliferation. Nutr Cancer 1998;31:111-8. [ Links ]
241. Fernandez-Barrena MG, Monte MJ, Latasa MU, Uriarte I, Vicente E, Chang HC, et al. Lack of Abcc3 expression impairs bile-acid induced liver growth and delays hepatic regeneration after partial hepatectomy in mice. J Hepatol 2012;56:367-73. [ Links ]
242. Chen WD, Wang YD, Zhang L, Shiah S, Wang M, Yang F, et al. Farnesoid X receptor alleviates age-related proliferation defects in regenerating mouse livers by activating forkhead box m1b transcription. Hepatology 2010;51:953-62. [ Links ]
243. Weymann A, Hartman E, Gazit V, Wang C, Glauber M, Turmelle Y, et al. P21 is required for dextrose-mediated inhibition of mouse liver regeneration. Hepatology 2009;50:207-15. [ Links ]
244. Fisette A, Hassanain M, Metrakos P, Doi SA, Salman A, Schricker T, et al. High-dose insulin therapy reduces postoperative liver dysfunction and complications in liver resection patients through reduced apoptosis and altered inflammation. J Clin Endocrinol Metab 2012;97:217-26. [ Links ]
245. Hassanain M, Metrakos P, Fisette A, Doi SA, Schricker T, Lattermann R, et al. Randomized clinical trial of the impact of insulin therapy on liver function in patients undergoing major liver resection. Br J Surg 2013;100:610-8. [ Links ]
246. Costa RH, Kalinichenko VV, Holterman AX, Wang X. Transcription factors in liver development, differentiation, and regeneration. Hepatology 2003;38:1331-47. [ Links ]
247. Wang X, Krupczak-Hollis K, Tan Y, Dennewitz MB, Adami GR, Costa RH. Increased hepatic Forkhead Box M1B (FoxM1B) levels in old-aged mice stimulated liver regeneration through diminished p27Kip1 protein levels and increased Cdc25B expression. J Biol Chem 2002;277:44310-6. [ Links ]
248. Brezillon N, Lambert-Blot M, Morosan S, Couton D, Mitchell C, Kremsdorf D, et al. Transplanted hepatocytes over-expressing FoxM1B efficiently repopulate chronically injured mouse liver independent of donor age. Mol Ther 2007;15:1710-5. [ Links ]
249. Marchioro TL, Porter KA, Brown BI, Otte JB, Starzl TE. The effect of partial portacaval transposition on the canine liver. Surgery 1967;61:723-32. [ Links ]
250. Starzl TE, Porter KA, Francavilla A. The Eck fistula in animals and humans. Curr Probl Surg 1983;20:687-752. [ Links ]
251. Starzl TE, Porter KA, Francavilla JA, Benichou J, Putnam CW. A hundred years of the hepatotrophic controversy. Ciba Found Symp 1977;(55):111-29. [ Links ]
252. Francavilla A, Starzl TE, Porter K, Foglieni CS, Michalopoulos GK, Carrieri G, et al. Screening for candidate hepatic growth factors by selective portal infusion after canine Eck's fistula. Hepatology 1991;14:665-70. [ Links ]
253. Gohda E, Tsubouchi H, Nakayama H, Hirono S, Sakiyama O, Takahashi K, et al. Purification and partial characterization of hepatocyte growth factor from plasma of a patient with fulminant hepatic failure. J Clin Invest 1988;81:414-9. [ Links ]
254. Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989;342:440-3. [ Links ]
255. Nakamura T, Sakai K, Nakamura T, Matsumoto K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J Gastroenterol Hepatol 2011;26 Suppl 1:188-202. [ Links ]
256. Michalopoulos G, Houck KA, Dolan ML, Leutteke NC. Control of hepatocyte replication by two serum factors. Cancer Res 1984;44:4414-9. [ Links ]
257. Noji S, Tashiro K, Koyama E, Nohno T, Ohyama K, Taniguchi S, et al. Expression of hepatocyte growth factor gene in endothelial and Kupffer cells of damaged rat livers, as revealed by in situ hybridization. Biochem Biophys Res Commun 1990;173:42-7. [ Links ]
258. Schirmacher P, Geerts A, Pietrangelo A, Dienes HP, Rogler CE. Hepatocyte growth factor/hepatopoietin A is expressed in fat-storing cells from rat liver but not myofibroblast-like cells derived from fat-storing cells. Hepatology 1992;15:5-11. [ Links ]
259. Block GD, Locker J, Bowen WC, Petersen BE, Katyal S, Strom SC, et al. Population expansion, clonal growth, and specific differentiation patterns in primary cultures of hepatocytes induced by HGF/SF, EGF and TGF alpha in a chemically defined (HGM) medium. J Cell Biol 1996;132:1133-49. [ Links ]
260. Rubin JS, Chan AM, Bottaro DP, Burgess WH, Taylor WG, Cech AC, et al. A broad-spectrum human lung fibroblast-derived mitogen is a variant of hepatocyte growth factor. Proc Natl Acad Sci U S A 1991;88:415-9. [ Links ]
261. Hernandez J, Zarnegar R, Michalopoulos GK. Characterization of the effects of human placental HGF on rat hepatocytes. J Cell Physiol 1992;150:116-21. [ Links ]
262. Mars WM, Liu ML, Kitson RP, Goldfarb RH, Gabauer MK, Michalopoulos GK. Immediate early detection of urokinase receptor after partial hepatectomy and its implications for initiation of liver regeneration. Hepatology 1995;21:1695-701. [ Links ]
263. Hamanoue M, Kawaida K, Takao S, Shimazu H, Noji S, Matsumoto K, et al. Rapid and marked induction of hepatocyte growth factor during liver regeneration after ischemic or crush injury. Hepatology 1992;16:1485-92. [ Links ]
264. Matsumoto K, Nakamura T. Emerging multipotent aspects of hepatocyte growth factor. J Biochem 1996;119:591-600. [ Links ]
265. Matsumoto K, Nakamura T. Hepatocyte growth factor (HGF) as a tissue organizer for organogenesis and regeneration. Biochem Biophys Res Commun 1997;239:639-44. [ Links ]
266. Benvenuti S, Comoglio PM. The MET receptor tyrosine kinase in invasion and metastasis. J Cell Physiol 2007;213:316-25. [ Links ]
267. Comoglio PM. Pathway specificity for Met signalling. Nat Cell Biol 2001;3:E161-2. [ Links ]
268. Stolz DB, Mars WM, Petersen BE, Kim TH, Michalopoulos GK. Growth factor signal transduction immediately after two-thirds partial hepatectomy in the rat. Cancer Res 1999;59:3954-60. [ Links ]
269. Lindroos PM, Zarnegar R, Michalopoulos GK. Hepatocyte growth factor (hepatopoietin A) rapidly increases in plasma before DNA synthesis and liver regeneration stimulated by partial hepatectomy and carbon tetrachloride administration. Hepatology 1991;13:743-50. [ Links ]
270. Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995;373:699-702. [ Links ]
271. Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A 2004;101:4477-82. [ Links ]
272. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000;103:211-25. [ Links ]
273. Sibilia M, Wagner EF. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995;269:234-8. [ Links ]
274. Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A 2007;104:17081-6. [ Links ]
275. Kiso S, Kawata S, Tamura S, Inui Y, Yoshida Y, Sawai Y, et al. Liver regeneration in heparin-binding EGF-like growth factor transgenic mice after partial hepatectomy. Gastroenterology 2003;124:701-7. [ Links ]
276. Berasain C, Garcia-Trevijano ER, Castillo J, Erroba E, Lee DC, Prieto J, et al. Amphiregulin: An early trigger of liver regeneration in mice. Gastroenterology 2005;128:424-32. [ Links ]
277. Loyer P, Cariou S, Glaise D, Bilodeau M, Baffet G, Guguen-Guillouzo C. Growth factor dependence of progression through G1 and S phases of adult rat hepatocytes in vitro. Evidence of a mitogen restriction point in mid-late G1. J Biol Chem 1996;271:11484-92. [ Links ]
278. Loyer P, Corlu A, Desdouets C. Regulation of the hepatocyte cell cycle: Signaling pathways and protein kinases. Int J Hepatol 2012;2012:592354. [ Links ]
279. Russell WE, Kaufmann WK, Sitaric S, Luetteke NC, Lee DC. Liver regeneration and hepatocarcinogenesis in transforming growth factor-alpha-targeted mice. Mol Carcinog 1996;15:183-9. [ Links ]
280. Webber EM, Wu JC, Wang L, Merlino G, Fausto N. Overexpression of transforming growth factor-alpha causes liver enlargement and increased hepatocyte proliferation in transgenic mice. Am J Pathol 1994;145:398-408. [ Links ]
281. Russell WE, Dempsey PJ, Sitaric S, Peck AJ, Coffey RJ, Jr. Transforming growth factor-alpha (TGF alpha) concentrations increase in regenerating rat liver: Evidence for a delayed accumulation of mature TGF alpha. Endocrinology 1993;133:1731-8. [ Links ]
282. Campbell JS, Prichard L, Schaper F, Schmitz J, Stephenson-Famy A, Rosenfeld ME, et al. Expression of suppressors of cytokine signaling during liver regeneration. J Clin Invest 2001;107:1285-92. [ Links ]
283. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003;113:685-700. [ Links ]
284. Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development 2009;136:3699-714. [ Links ]
285. Brown KA, Pietenpol JA, Moses HL. A tale of two proteins: Differential roles and regulation of Smad2 and Smad3 in TGF-beta signaling. J Cell Biochem 2007;101:9-33. [ Links ]
286. Chari RS, Price DT, Sue SR, Meyers WC, Jirtle RL. Down-regulation of transforming growth factor beta receptor type I, II, and III during liver regeneration. Am J Surg 1995;169:126,31; discussion 131-2. [ Links ]
287. Macias-Silva M, Li W, Leu JI, Crissey MA, Taub R. Up-regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor-beta signals during liver regeneration. J Biol Chem 2002;277:28483-90. [ Links ]
288. Nishikawa Y, Wang M, Carr BI. Changes in TGF-beta receptors of rat hepatocytes during primary culture and liver regeneration: increased expression of TGF-beta receptors associated with increased sensitivity to TGF-beta-mediated growth inhibition. J Cell Physiol 1998;176:612-23. [ Links ]
289. Russell WE, Coffey RJ,Jr, Ouellette AJ, Moses HL. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc Natl Acad Sci U S A 1988;85:5126-30. [ Links ]
290. Bouzahzah B, Fu M, Iavarone A, Factor VM, Thorgeirsson SS, Pestell RG. Transforming growth factor-beta1 recruits histone deacetylase 1 to a p130 repressor complex in transgenic mice in vivo. Cancer Res 2000;60:4531-7. [ Links ]
291. Oe S, Lemmer ER, Conner EA, Factor VM, Leveen P, Larsson J, et al. Intact signaling by transforming growth factor beta is not required for termination of liver regeneration in mice. Hepatology 2004;40:1098-105. [ Links ]
292. Kogure K, Zhang YQ, Maeshima A, Suzuki K, Kuwano H, Kojima I. The role of activin and transforming growth factor-beta in the regulation of organ mass in the rat liver. Hepatology 2000;31:916-21. [ Links ]
293. Uygun BE, Soto-Gutierrez A, Yagi H, Izamis ML, Guzzardi MA, Shulman C, et al. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat Med 2010;16:814-20. [ Links ]
294. Uygun BE, Yarmush ML, Uygun K. Application of whole-organ tissue engineering in hepatology. Nat Rev Gastroenterol Hepatol 2012;9:738-44. [ Links ]
295. Soto-Gutierrez A, Wertheim JA, Ott HC, Gilbert TW. Perspectives on whole-organ assembly: Moving toward transplantation on demand. J Clin Invest 2012;122:3817-23. [ Links ]
296. Hammond JS, Gilbert TW, Howard D, Zaitoun A, Michalopoulos G, Shakesheff KM, et al. Scaffolds containing growth factors and extracellular matrix induce hepatocyte proliferation and cell migration in normal and regenerating rat liver. J Hepatol 2011;54:279-87. [ Links ]
297. Badylak SF, Taylor D, Uygun K. Whole-organ tissue engineering: Decellularization and recellularization of three-dimensional matrix scaffolds. Annu Rev Biomed Eng 2011;13:27-53. [ Links ]
298. Baptista PM, Siddiqui MM, Lozier G, Rodriguez SR, Atala A, Soker S. The use of whole organ decellularization for the generation of a vascularized liver organoid. Hepatology 2011;53:604-17. [ Links ]
299. Gkretsi V, Bowen WC, Yang Y, Wu C, Michalopoulos GK. Integrin-linked kinase is involved in matrix-induced hepatocyte differentiation. Biochem Biophys Res Commun 2007;353:638-43. [ Links ]
300. Gkretsi V, Apte U, Mars WM, Bowen WC, Luo JH, Yang Y, et al. Liver-specific ablation of integrin-linked kinase in mice results in abnormal histology, enhanced cell proliferation, and hepatomegaly. Hepatology 2008;48:1932-41. [ Links ]
301. Apte U, Gkretsi V, Bowen WC, Mars WM, Luo JH, Donthamsetty S, et al. Enhanced liver regeneration following changes induced by hepatocyte-specific genetic ablation of integrin-linked kinase. Hepatology 2009;50:844-51. [ Links ]
302. Shafiei MS, Rockey DC. The role of integrin-linked kinase in liver wound healing. J Biol Chem 2006;281:24863-72. [ Links ]
303. Song JJ, Guyette JP, Gilpin SE, Gonzalez G, Vacanti JP, Ott HC. Regeneration and experimental orthotopic transplantation of a bioengineered kidney. Nat Med 2013;19:646-51. [ Links ]
304. Song JJ, Kim SS, Liu Z, Madsen JC, Mathisen DJ, Vacanti JP, et al. Enhanced in vivo function of bioartificial lungs in rats. Ann Thorac Surg 2011;92:998,1005; discussion 1005-6. [ Links ]
305. Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell 2004;118:9-17. [ Links ]
306. Ben-Porath I, Weinberg RA. When cells get stressed: An integrative view of cellular senescence. J Clin Invest 2004;113:8-13. [ Links ]
307. Morgan DO. The DNA damage response. In: Morgan DO, editor. The cell cycle: Principles of control. London: New Science Press; 2007. p. 227-45. [ Links ]
308. Campisi J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005;120:513-22. [ Links ]
309. Weinberg RA. P53 and apoptosis: master guardian and executioner. In: Weinberg RA, editor. The biology of cancer. New York: Garland Science; 2007. p. 307-56. [ Links ]
310. Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2002;2:594-604. [ Links ]
311. Levine AJ. P53, the Cellular Gatekeeper for Growth and Division. Cell 1997;88:323-31. [ Links ]
312. Efeyan A, Serrano M. P53: Guardian of the genome and policeman of the oncogenes. Cell Cycle 2007;6:1006-10. [ Links ]
313. Li CC, Chu HY, Yang CW, Chou CK, Tsai TF. Aurora-A overexpression in mouse liver causes p53-dependent premitotic arrest during liver regeneration. Mol Cancer Res 2009;7:678-88. [ Links ]
314. Maddocks OD, Vousden KH. Metabolic regulation by p53. J Mol Med (Berl) 2011;89:237-45. [ Links ]
315. 413.Fededa JP, Gerlich DW. Molecular control of animal cell cytokinesis. Nat Cell Biol 2012;14:440-7. [ Links ]
316. Gallagher SJ, Kefford RF, Rizos H. The ARF tumour suppressor. Int J Biochem Cell Biol 2006;38:1637-41. [ Links ]
317. Wadhwa R, Sugihara T, Taira K, Kaul SC. The ARF-p53 senescence pathway in mouse and human cells. Histol Histopathol 2004;19:311-6. [ Links ]
318. Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett 2006;237:10-21. [ Links ]
319. Demetris AJ, Seaberg EC, Wennerberg A, Ionellie J, Michalopoulos G. Ductular reaction after submassive necrosis in humans. Special emphasis on analysis of ductular hepatocytes. Am J Pathol 1996;149:439-48. [ Links ]
320. Stravitz RT, Kramer D J. Acute liver failure. En: Boyer TD, Manns MP, Sanyal AJ, editores. Zakim and Boyer's hepatology: a textbook of liver disease. 6th ed. Philadelphia: Saunders Elsevier; 2012. p. 327-51. [ Links ]
321. Oda M, Yokomori H, Han JY. Regulatory mechanisms of hepatic microcirculatory hemodynamics: hepatic arterial system. Clin Hemorheol Microcirc 2006;34:11-26. [ Links ]
322. Ninomiya M, Shirabe K, Terashi T, Ijichi H, Yonemura Y, Harada N, et al. Deceleration of regenerative response improves the outcome of rat with massive hepatectomy. Am J Transplant 2010;10:1580-7. [ Links ]
323. Markiewski MM, DeAngelis RA, Lambris JD. Liver inflammation and regeneration: Two distinct biological phenomena or parallel pathophysiologic processes? Mol Immunol 2006;43:45-56. [ Links ]
324. Clavien PA, Oberkofler CE, Raptis DA, Lehmann K, Rickenbacher A, El-Badry AM. What is critical for liver surgery and partial liver transplantation: Size or quality? Hepatology 2010;52:715-29. [ Links ]
325. Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013;499:481-4. [ Links ]
326. Ordonez MP, Goldstein LS. Using human-induced pluripotent stem cells to model monogenic metabolic disorders of the liver. Semin Liver Dis 2012;32:298-306. [ Links ]
327. Beginner´s Traps. In: Ramón y Cajal S, editor. Advice for a young investigator. Cambridge, Massachussetts: Massachussetts Institute of Technology; 1999. p. 9-27. [ Links ]

