










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.109 no.2 Madrid feb. 2017
https://dx.doi.org/10.17235/reed.2016.4224/2016
Management of pancreatic gastrinoma associated with Von Hippel-Lindau disease: a case report
Manejo del gastrinoma pancreático asociado a la enfermedad de Von Hippel-Lindau. A propósito de un caso
Ángela Sala-Hernández, Eva María Montalvá-Orón, Eugenia Pareja-Ibars, Neus Ballester-Pla and Rafael López-Andújar
Department of General Surgery. Hospital Universitario Politécnico La Fe. Valencia, Spain
ABSTRACT
Background: Pancreatic neuroendocrine tumors (PNET) are a heterogeneous group and constitute 1.3% of all pancreatic tumors. Approximately 10% of these occur in the context of hereditary syndromes, such as VHL disease.
Case report: We report a case of a female patient of 37 years diagnosed VHL and intervened on several occasions by cerebral hemangioblastoma and renal carcinomas. During its follow-up she was diagnosed 2 gastrinomas functioning under 2 cm were enucleated. Later developed new PNET and underwent a total duodenopancreatectomy without pyloric preservation.
Discussion: The management of PNET in VHL is difficult due to the association of multiple tumors in different organs and the morbidity and mortality associated with the surgery of the pancreas. Management must be individualized for each patient, based on the ability to produce hormones and present symptoms, the size and location, and in the context of other tumors that usually present in these patients.
Key words: Von Hippel-Lindau. Neuroendocrine tumour. Pancreas. Gastrinoma.
RESUMEN
Introducción: los tumores neuroendocrinos de páncreas (TNEP) son un grupo heterogéneo y constituyen el 1,3% de todos los tumores pancreáticos. Aproximadamente el 10% aparecen en el contexto de síndromes familiares como el Von Hippel-Lindau (VHL).
Caso clínico: presentamos el caso de una paciente mujer de 37 años diagnosticada de VHL e intervenida en varias ocasiones por hemangioblastomas cerebrales y carcinomas renales. Durante su seguimiento se diagnostica de 2 gastrinomas funcionantes menores de 2 cm que se enuclearon. Posteriormente desarrolló nuevo TNEP y se le realizó una duodenopancreatectomía total sin preservación pilórica.
Discusión: el manejo de los TNEP en el VHL es difícil debido a la asociación de múltiples tumores en diferentes órganos y a la morbi-mortalidad asociada a la cirugía del páncreas. Su tratamiento hay que individualizarlo en cada paciente, basándonos en su capacidad de producción de hormonas y, por tanto de dar sintomatología, en su tamaño y localización y, además debe ser contextualizado con el resto de tumores que suelen presentar estos pacientes.
Palabras clave: Von Hippel-Lindau. Tumor neuroendocrino. Páncreas. Gastrinoma.
Introduction
Von Hippel-Lindau (VHL) disease is rare. It has an autosomal dominant pattern of inheritance, and is characterized by the development of tumors and cysts in various organs.
Tumors most frequently appear in the following organs: kidney (renal clear cell carcinoma) (25-60%), adrenal gland (pheochromocytoma) (10-20%), central nervous system (haemangioblastoma) (44-72%), eye (retinal angioma) (25-60%), inner ear (endolymphatic sac neoplasia) (10-25%), epididymis (cystadenoma) (25-60%), and pancreas (neuroendocrine tumors and cysts) (35-77%) (1,2).
Serous cysts are the most common lesion in the pancreas (70%) (3), while neuroendocrine tumors appear in 10-17% of cases (4), which are usually unique and non-functioning.
Unlike sporadic pancreatic neuroendocrine tumors, which are usually non-functioning, sporadic, and have a high potential for malignancy (60-100%), pancreatic neuroendocrine tumors (PNET) related to VHL disease have a lower rate of metastasis. Because they do not secrete hormones, they produce no symptoms and are therefore usually diagnosed in advanced stages. Usually, patients with VHL disease undergo screening programs from a young age, allowing early diagnosis of PNET (5).
This case report presents a case of a functioning and malignant PNET in a patient with VHL disease and a multi-cystic pancreas.
Case report
A 29-year-old female patient with VHL disease, with a ventriculoperitoneal shunt. She underwent two interventions for renal cell carcinoma (right kidney partial resection and radiofrequency of a nodule in the left kidney), and four resections of cerebral hemangioblastoma.
During follow-up, positron emission tomography-computed tomography (PET-CT) detected two pancreatic head lesions with a maximum standardized uptake value (SUV max) of 14.94 g/mL and multiple pancreatic cysts (Fig. 1). Next, a pancreatic functional study was performed, and revealed a high level of serum gastrin (2,319 pg/ml). Transgastric fine needle aspiration (FNA) yielded findings compatible with pancreatic neuroendocrine tumor.

With the diagnosis of two non-functioning gastrinomas sized less than 2 cm (T1N0M0, stage IV, differentiation grade G2, less than 2 mitoses per field, and Ki67 2-5%), a multidisciplinary committee decided that conservative surgery was indicated (6).
At surgery, after wide exposure of the pancreas, intraoperative ultrasound localized the two solid lesions at the pancreatic head and uncinate process, and enucleation with an ultrasonic dissector (CUSA) and harmonic scalping were performed. The postoperative course was uneventful.
Anatomopathological assessment revealed two moderately differentiated neuroendocrine tumors (G2) (chromogranin, synaptophysin and CD56+), with an index cell proliferation (Ki67) of 10% and atypical mitotic figures (1 mitosis in 10 high power fields). Postoperative gastrin level was significantly decreased, but still out of the normal range (1,383 pg/ml).
At 7 months follow-up, a new elevation of this biomarker was detected, as well as a high level of chromogranin A (181 ng/ml).
Although the Octreoscan did not reveal abnormal accumulation of activity, magnetic resonance (RM) and PET-TC showed growth of the left kidney tumor previously treated with radiofrequency and a new lesion in the pancreatic head in contact with the superior mesenteric vein (Fig. 2).
Given these findings, and in the context of a multi-cystic pancreas, the multidisciplinary committee decided to perform a total pancreaticoduodenectomy without pyloric preservation and associated splenectomy, with partial left kidney resection. To reconstruct bowel continuity, an end-to-side transmesocolic gastrojejunostomy and an end-to-side hepaticojejunostomy Roux-en-Y were performed. It was necessary to perform a partial portal resection because it was not possible to identify a separation plane between the portal vein and pancreas. The portal vein was reconstructed with a continuous suture. The intraoperative biopsy ruled out vessel tumor infiltration (Fig. 3).
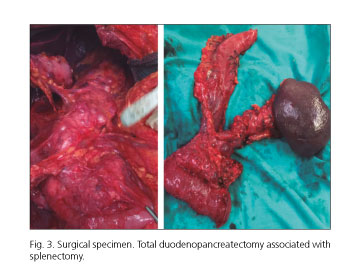
The histological report described a 9-mm pancreatic neuroendocrine tumor in the uncinate process, with moderate differentiation and without lymph node infiltration (0/8), and multiple cystic formations in the pancreas (multifocal and multicystic serous cystadenomas, with areas of serous-solid adenomas), and a 28-mm renal cell carcinoma.
During the postoperative course, the patient presented a caliceal urinary fistula requiring drainage by interventional radiology, which resolved after 2 months of follow-up (grade III complication according to the Clavien-Dindo classification).
Discussion
Pancreatic neuroendocrine tumors are a heterogeneous group, representing 1.3% of all pancreatic tumors.
Approximately 10% of these occur in the context of hereditary syndromes, such as VHL disease. PNET in patients with VHL disease has a better prognosis than sporadic PNET (10-20% and 60-90% of metastasic diseases, respectively).
These neoplasms may exhibit malignant behavior (5,7). They can be functioning or non-functioning. While the former clinically manifest in relation to secreted hormones, the latter usually have symptoms secondary to extrinsecal compression of neighboring structures, or are detected as incidentalomas during examinations performed because of other diseases.
Hormonal, imaging, and histopathological tests must be performed to evaluate and diagnose PNET.
It is vital to assess the plasma levels of biochemical markers when diagnosing and monitoring patients with functioning neuroendocrine tumors.
In this case, a functional study revealed elevated gastrin levels, which dropped after the first surgery and increased again in subsequent controls, which helped in the detection of new recurrence.
Conventional imaging studies for PNET localization include endoscopic ultrasound, TC, and MRI. Radioactively labeled ligands are now available; these have led to major advances in the diagnosis, localization, and follow-up of these tumors. Octreotide scintigraphy and PET-TC (5) are complementary tests available for localization of these lesions. In this patient, the location was determined using PET-TC, whereas the Octreoscan results were found to be a false negative.
Managing PNET in patients with VHL disease is challenging because of the association of multiple tumors in different organs and the morbidity and mortality associated with surgery of the pancreas.
For asymptomatic tumors, some authors rely on size and location of the lesion for management, recommending follow-up with contrast-TC or RM every 12 months when lesions are smaller than 1 cm. In lesions sized 1-3 cm, management depends on location: surgical resection is indicated for tumors larger than 2 cm when located in the head of the pancreas, and in tumors larger than 3 cm when located in the body of the pancreas (4).
For functioning tumors, the recommended treatment is surgical resection regardless of size and location (2,9).
Blansfield et al. (4) proposed management depending on the likelihood of metastasic disease based on three factors: the presence of mutation in exon 3, size duplication time of less than 500 days, and size larger than 3 cm. Patients who have none of these indicators are at little risk for metastatic disease and radiological follow-up is recommended every 2-3 years; if they have one of these indicators, monitoring is recommended every 6-12 months; and if they have two or three of these indicators, they are at high risk for metastatic disease and surgical resection is recommended (10).
Surgical treatment options range from enucleation, for small or low-grade tumors away from the pancreatic duct, to partial or total resection (9).
Surgery is the recommended management in case of tumor recurrence, as in this patient, when the tumor is able to be completely resected.
Conclusion
Functioning neuroendocrine pancreatic tumors associated with VHL disease are rare. Management must be individualized for each patient, based on the ability to produce hormones and present symptoms, the size and location, and in the context of other tumors that usually present in these patients.
References
1. Eras M1, Yenigun M, Acar C, et al. Pancreatic involvement in Von Hippel-Lindau disease. Indian J Cancer 2004; 41:159-61. [ Links ]
2. Lonser RR, Glenn GM, Walther M, et al. Von Hippel-Lindau disease. Lancet 2003;361:2059-67. DOI: 10.1016/S0140-6736(03)13643-4. [ Links ]
3. Hammel PR, Vilgrain V, Terris B, et al.Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d'Etude de la Maladie de von Hippel-Lindau. Gastroenterology 2000;119:1087-95. DOI: 10.1053/gast.2000.18143. [ Links ]
4. Blansfield JA, Choyke L, Morita SY, et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine tumors. Surgery 2007;142:814-8. DOI: 10.1016/j.surg.2007.09.012. [ Links ]
5. Ro C1, Chai W, Yu VE, et al. Pancreatic neuroendocrine tumors: biology, diagnosis, and treatment. Chin J Cancer 2013;32:312-24. DOI: 10.5732/cjc.012.10295. [ Links ]
6. Klöppel G, Rindi G, Perren A, et al. The ENETS and AJCC/UICC TNM classifications of the neuroendocrine tumors of the gastrointestinal tract and the pancreas: a statement. Virchows Arch 2010;456(6):595-7. [ Links ]
7. Libutti SK, Choyke PL, Bartlett DL, et al. Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recommendations. Surgery 1998;124:1153-9. DOI: 10.1067/msy.1998.91823. [ Links ]
8. Tan EH, Tan CH. Imaging of gastroenteropancreatic neuroendocrine tumors. World J Clin Oncol 2011;2:28-43. DOI: 10.5306/wjco.v2.i1.28. [ Links ]
9. Charlesworth M, Verbeke CS, Falk GA, et al. Pancreatic lesions in von Hippel-Lindau disease? A systematic review and meta-synthesis of the literature.J Gastrointest Surg 2012;16:1422-8. DOI: 10.1007/s11605-012-1847-0. [ Links ]
10. Kulke MH, Shah MH, Benson AB 3rd. Neuroendocrine tumors, version 1.2015. J Natl Compr Canc Netw 2015;13:78-108. [ Links ]
![]() Correspondence:
Correspondence:
Ángela Sala-Hernández.
Department of General Surgery.
Hospital Universitario Politécnico La Fe.
Avda. de Fernando Abril Martorell, 106.
46026 Valencia, Spain
e-mail: asalahdez@gmail.com
Received: 29-01-2016
Accepted: 25-02-2016
