










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.109 n.6 Madrid Jun. 2017
https://dx.doi.org/10.17235/reed.2017.4228/2016
ORIGINAL PAPERS
Post-transplant lymphoproliferative disease in liver transplant recipients
Síndrome linfoproliferativo en el trasplante hepático
Mercedes Rubio-Manzanares-Dorado, José María Álamo-Martínez, Carmen Bernal-Bellido, Luis Miguel Marín-Gómez, Gonzalo Suárez-Artacho, Carmen Cepeda-Franco, Jize Wang, Miquel Ángel Gómez-Bravo and Javier Padillo
Unit of Hepatobiliary Surgery and Liver Transplantation. Hospital Universitario Virgen del Rocío. Sevilla, Spain
ABSTRACT
Introduction: Post-transplant lymphoproliferative syndrome (PTLD) is a rare and potentially life-threatening complication after liver transplantation. The aim of this study was to analyze the clinicopathologic features related to PTLD in a single institution after liver transplantation.
Methods: Observational study where we have retrospectively analyzed 851 cases who underwent liver transplantation. Ten cases have developed PTLD. Their clinical-pathological characteristics and the treatment received have been analyzed.
Results: PTLD incidence was 1.2% (10/851). The mean time from liver transplantation to PTLD diagnosis was 36 months (range 1.2 to 144 months). PTLD localization was extranodal in all cases, the most frequent location being intestinal. Seven cases showed a monomorphic lymphoma which in all cases was differentiated B cell lymphomas. Fifty per cent of the series were seropositive for Epstein-Barr virus. Five patients were alive at the time of the review. Among these patients, we observed three cases of complete remission and two cases of disease stabilization. The death rate was higher in the first year after diagnosis of PTLD.
Conclusion: PTLD is a rare complication after liver transplantation, but it may pose a threat to the life of a liver transplant recipient. It is essential to identify patients at risk, to establish an early diagnosis and treatment that can change the outcome of the disease.
Key words: Post-transplant lymphoproliferative disease. Liver transplantation. Rituximab.
RESUMEN
Introducción: el síndrome linfoproliferativo postrasplante (SLPT) es una complicación infrecuente que ensombrece el pronóstico de los pacientes sometidos a un trasplante hepático (TH). Su patogenia es multifactorial, siendo sus dos principales factores de riesgo la inmunodepresión y la infección del virus de Epstein-Barr (VEB); sin embargo, en actualidad se piensa que puede estar relacionada con otros factores.
Métodos: estudio observacional en el que hemos analizado de forma retrospectiva 851 casos que fueron sometidos a un trasplante hepático, de los cuales diez casos han desarrollado un SLPT. Se han analizado sus características clinicopatológicas y el tratamiento recibido.
Resultados: la incidencia del SLPT ha sido del 1,2% (10/851) y el tiempo medio de presentación desde el TH hasta el diagnóstico, de 36 meses (rango 1,2-144 meses). El lugar de presentación ha sido extranodal en todos los casos, siendo más frecuente la localización intestinal. Siete casos presentaron un SLPT monomorfo, todos ellos linfomas diferenciados de células B. El 50% de la serie presentó seronegatividad para el virus de Epstein-Barr. La supervivencia global ha sido del 50%. Entre estos pacientes, hemos observado tres casos de curación completa, un caso de estabilización de la enfermedad y otro caso de recurrencia.
Conclusión: el SLPT es una complicación infrecuente que supone una amenaza para la vida del paciente. Para poder instaurar un diagnóstico precoz y un tratamiento que pueda modificar el curso de la enfermedad, es fundamental la identificación de los pacientes en riesgo.
Palabras clave: Síndrome linfoproliferativo postrasplante. Trasplante hepático. Rituximab.
Introduction
During the past 20 years, liver transplantation programs have undergone a huge development. Advances in preoperative management, improvements in surgical techniques and progress in postoperative care have facilitated the improvement of liver transplant patient survival rates. The survival results published in European and American records exceed 85%, 75% and 60% at one, five and ten years respectively (1).
However, long-term complications are a major cause overshadowing the prognosis of these patients. It has been observed that patients with solid organ transplantation are at increased risk of developing malignancies (2). Several factors have been described to contribute to the development of tumors. The reduction of immune surveillance mechanisms in the recipient, the activation of latent virus with oncological potential and the chronic use of immunosuppressive agents are the main factors for the appearance of cancer after a solid organ transplantation. In our country, the incidence of malignancies in liver transplantation is about 8%. The most common are skin tumors, followed by lymphoproliferative disorders (3).
Post-transplant lymphoproliferative disease (PTLD) is a rare but also a serious complication in solid organ transplantation. In liver transplantation recipients, PTLD is reported in up to 1 to 2.8% of adults and 15% of children (4). It has been closely associated with exogenous immunosuppression and the Epstein-Barr virus (EBV); in fact, EBV is associated in up to 90% of cases. PTLD mortality has been estimated at around 50%, although progress in treatment and chemotherapy regimens appear to have improved these results (5-9). Despite these different treatment options, mortality rates remain high. For this reason, early diagnosis is essential to facilitate prompt treatment.
Relatively little is known about the incidence and outcome of PTLD in adults with a transplanted liver. The main objective of this study was to analyze the clinical-pathological characteristics of one of the most uncommon complications after liver transplantation.
Material and methods
In this observational study, 851 cases who underwent liver transplantation in the Hospital Universitario Virgen del Rocío (Seville, Spain) from 1994 to 2011 were analyzed. Among the 851 cases analyzed, ten patients developed a PTLD that was diagnosed in all cases by biopsy of nodal or extranodal sites detected on imaging tests. Immunohistochemical markers were used to delineate the subtype of lymphoid proliferation, including CD20, CD79a, CD3, CD10 and bcl-6. Lymphomas were classified according to the World Health Organization (WHO) classification of PTLD (Table I).

Inclusion and exclusion criteria
We have included in this study all patients who received an isolated liver transplantation (not split or living donor) and developed PTLD. We have excluded from the study those who did not meet PTLD features according the WHO classification (Table I).
Variables analysed
The variables included in the study were as follows: age, gender, cause for liver transplantation, immunosuppressive agents used at the time of diagnosis, presence of acute rejection and need for treatment, lactate dehydrogenase (LDH) > 250 mg/dl before PTLD detection, PTLD histological classification, agents used after the PTLD diagnosis, chemotherapy used and use of rituximab.
EBV was diagnosed before the transplantation both in the donor and in the recipient by RNA in situ hybridization.
Hepatitis C virus (HCV) recurrence was diagnosed by biopsy on the new liver graft prior to the development of PTLD, and confirmed by the HCV RNA detection.
Immunosuppression was based on triple therapy, consisting of calcineurin inhibitor (tacrolimus [FK 506]/cyclosporin A [CyA]), steroids and mycophenolate mofetil/azathioprine. Corticosteroids were removed from the third month.
We collected the cases that had a history of treated acute cellular rejection episodes. These cases were treated with 6-methylprednisolone intravenous bolus. In the same way, we have also collected patients who received induction therapy with OKT3 or anti-CD25 antibodies.
Finally, we have analyzed those cases that converted from a calcineurin agent to mTOR inhibitors (sirolimus/everolimus) after diagnosis of PTLD.
Evaluation of response to treatment and survival
Treatment response was assessed using follow-up computed tomographic (CT) scans. Restaging was performed following treatment. Complete remission was defined as the disappearance of all clinical evidence of active tumor for at least one month. Partial remission was defined as a minimum of 50% reduction in the measurement of detectable lesions for at least one month. Stable disease was considered when no objective modifications in the lesions were observed. Progressive disease was defined as the enlargement of tumor size or the occurrence of new lesions.
Survival was defined as days survived from the date of diagnosis of PTLD to date of death or until the date of last revision.
Statistical analysis
Demographic characteristics, clinical presentation, histology and survival data were collected retrospectively in July 2013 in a SPSS 15.0 database (Tables II and III). A descriptive analysis was performed.

For quantitative variables, data were expressed using means ± standard derivation, and percentage and counts were used for qualitative variables. For the analysis of two qualitative variables in a sample of unequal size we used the Fisher's exact test. Statistical significance was considered if p < 0.05.
Finally, the Kaplan-Meier method was used to estimate survival following PTLD diagnosis.
Results
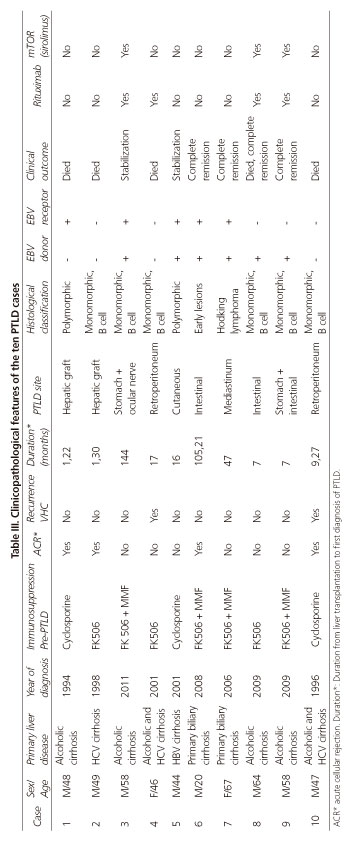
During the study period (1994-2011), the incidence of proven PTLD was 1.2% (10/851). Of the ten patients with histological PTLD, eight were male (80%) and two were women (20%). All patients studied were adults over 18 years of age. The mean age at disease presentation was 50 years (range 20-67). The causes underlying the liver transplantation included four patients with alcoholic cirrhosis, one case with hepatitis C cirrhosis, one case with hepatitis B cirrhosis and two cases of primary biliary cirrhosis. In addition, two patients had mixed alcohol cirrhosis and hepatitis C cirrhosis (Tables II and III).
The timing from liver transplantation to first diagnosis of PTLD mean was two years and nine months (range 12 months to 12 years). In 50% of cases, PTLD was diagnosed during the first year after liver transplantation, whereas 30% presented a late onset, within three years after transplantation.
Regarding clinical presentation, although symptoms were varied, the most common manifestations were fever and epigastric pain (30%). Other presentations were one case of intestinal obstruction, two cases of skin lesions, two cases of anemia, and one patient had chylous ascites. One case presented as a graft acute cellular rejection (Tables II and III).
The most common manifestation in clinical examination and imaging tests was as a tumor mass. PTLD localization was extranodal in all cases, the most frequent location was intestinal. Among the four cases of intestinal localization, two were located in the stomach and two in the small intestine (one of these patients had both sites). Other sites include two retroperitoneal masses, one mediastinal mass and a cutaneous lymphoma. In two patients the PTLD was located on the liver graft, although no evidence of tumor mass in the previous image was detected.
Following the WHO classification of PTLD (Table I), one patient had early lesions showing plasmacytic hyperplasia features, two patients were polymorphic PTLD, whereas seven cases remained monomorphic. Among the latter, six patients were B cell lymphomas (five resembling diffuse large B cell lymphoma) and one patient was Hodgkin lymphoma. No patient had monomorphic T cell lymphoma.
The status for the Epstein-Barr virus (EBV) receptor was performed in all cases by in situ RNA hybridization for EBV. Fifty per cent of patients were EBV positive and the remaining 50% were negative. All patients who had negative serology for EBV showed a monomorphic B cell lymphoma.
Regarding the EBV status in the donor liver, 60% presented seropositivity for EBV. Among them, two grafts with positivity for EBV were implanted in recipients with negative EBV. In our series we found no significant difference among patients with EBV seronegativity who received a liver from a seropositive donor from those who received a negative EBV graft (Fisher's exact test p = 1).
Concerning HCV recurrence in the graft after liver transplantation, two of the three patients transplanted for virus C cirrhosis had a recurrence of the disease in the graft. Lymphoma appearance was earlier in these cases. Both patients were monomorphic B cell lymphomas (Table III).
LDH levels were determined prior to diagnosis of PTLD in eight cases, observing an elevation above 250 mg/dl in seven of them.
Seven patients have been treated with tacrolimus as a basic immunosuppressive therapy. Among these cases, five patients developed a B cell lymphoma, one case was a Hodgkin lymphoma and another case was plasmocytic hyperplasia. Among the three patients who received cyclosporine, two developed a polymorphic lymphoma and one, a monomorphic B cell lymphoma.
Although there is a tendency to use lower immunosuppressive levels in the latter years of the study, we have not seen a lower number of cases in this period. Fifty percent of the cases were diagnosed in the latter half of the study (Table III).
Four patients required intravenous treatment with 6-methylprednisolone (6-MP) for acute rejection. Among them, three patients received OKT3. In these cases, PTLD developed earlier.
Three patients were diagnosed post mortem (30%) and they were not treated. The immunosuppression reduction without any other treatment allowed the achievement of complete remission of early lesions in one case and the stabilization of a cutaneous lymphoma in another. Three patients received rituximab as a first-line treatment and one patient as a second-line therapy. The latter received rituximab when current protocols for the administration of this drug were not established, thus receiving rituximab after chemotherapy. Immunosuppressive therapy of the three cases that received rituximab as first-line treatment was substituted for an mTOR inhibitor after diagnosis of PTLD. Specifically, these patients received rapamycin (Sirolimus®) plus mycophenolate mofetil, among which a complete cure has been observed in two cases and disease stabilization in the remaining case.
Two patients received chemotherapy. Among them, the patient who received rituximab as second-line treatment died. The other case was a Hodgkin lymphoma who received chemotherapy with ABVD regimen as first-line and CEP as second-line treatment with a resulting complete remission.
The mortality in our series was 50%. The overall complete cure rate was 40%. One of the patients that had a complete remission died due to another cause not related with PTLD, specifically, he died due to a heart attack. We also observed a recurrence of 10%, resulting in death and rates of disease stabilization of 20%. The minimum follow-up time of patients from diagnosis of SLPT to the present time has been three years.
It should be noted that since the use of rituximab (four patients) and calcineurin conversion to mTOR inhibitors (three patients) the cure rate has been 66%, with one case of stabilization (33%). The latter patient had at the time of diagnosis a primary tumor in the stomach and a metastasis in the ocular nerve. After he received rituximab, the primary tumor completely disappeared but the metastasis still persisted. For this reason, he is waiting to receive radiotherapy.
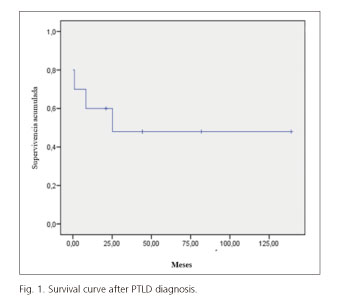
Survival mean was 71 ± 21 months and the actuarial survival at the time of the study was 50%. The overall survival at one, three and five years was 60%, 50% and 50%, respectively (Fig. 1). In three cases, the diagnosis of PTLD was made post mortem. In the remaining two cases, death occurred at eight and 25 months of diagnosis.
Discusion
The incidence of PTLD after liver transplantation has been reported to vary from 2% to 5% (3,4). In our series, incidence was largely in accordance with the reference range in the literature, specifically, it was in up to 1.2% of cases.
Although PTLD is a rare complication, it can be a threat to the patient's life due to its high mortality rate and rapid clinical progression. It is estimated that the mortality of PTLD is around 50% (5-9). In our series, the survival mean from diagnosis of PTLD to death or the last contact with the patient was 71 ± 21 months. We have observed that the survival rate after the first year of PTLD diagnosis was 60%. The death rate has been higher in the first year after diagnosis of PTLD.
The median time for presentation was 36 months (range 1.2 to 144 months). In 70% of cases the diagnosis was made in the two years following liver transplantation. These findings are similar to those found in the literature, where the highest incidence of PTLD is observed at 18 months (10).
Risk factors for PTLD development
Risk factors for PTLD after liver transplant include EBV seronegativity, early age (especially children and adolescents), and high doses of immunosuppression and first year after transplant (11-13). In our case, 50% of recipients had EBV seronegativity prior to the liver transplant. Although multiple risk factors for the development of PTLD have been reported, the two main factors are EBV seronegativity of the recipient and the degree of immunosuppression. Recipients with EBV seronegativity who received a liver from a seropositive donor are at a particularly high risk due to the lack of specific immunity (cytotoxic T lymphocytes) to fight against infected B cells (14). In our series, no significant difference was observed, and this may be due to the limited number of receptors with seronegativity for EBV (Fisher's exact test p = 1).
In the same way, the administration of high doses of steroids as a result of acute graft rejection has also been associated with an increased risk of PTLD (15). Immunosuppression after transplantation, leading to a decrease of the specific response function of cytotoxic T lymphocytes against EBV generates an uncontrolled proliferation mediated by the virus which culminates with the appearance of PTLD. In our series, 40% of patients had acute liver graft rejection. As treatment, all patients received intravenous steroids. Among them three cases were also treated with OKT3. The latter may have had a greater predisposition to the development of PTLD due to the high degree of immunosuppression.
Other risk factors that have been associated with PTLD are HCV recurrence on the graft, age older than 50 years, alcoholic cirrhosis and hepatitis C cirrhosis (16). In our series, all patients were of legal age, with an average age of presentation of 50 years (range 20-67 years). The principal causes which motivated liver transplantation were alcohol and hepatitis C virus. Among patients with HCV, we have observed recurrence of hepatitis C virus on the graft in half of the cases. Immunosuppression received after liver transplantation suppress immunosurveillance mechanisms facilitating the activation of the virus with oncogenic potential that can lead to the development of tumors.
PTLD diagnosis
Clinical presentation often includes fever, lymphadenopathy, weight loss and splenomegaly. The disease can affect a single organ, graft or multiple organs. Bone marrow failure and central nervous system dysfunction is also common (6,17). Although in our series the most common clinical manifestations were fever along with epigastric pain (30%), in fact, the symptoms were varied.
As noted, the first manifestations of PTLD are widely variable; for this reason, a high index of suspicion for early detection of PTLD is required. To resolve this, the measurement of EBV load by quantitative polymerase chain reaction can be an important weapon for monitoring and diagnosing patients with PTLD risk factors. Although a high load of EBV has a high sensitivity to predict PTLD, specificity in the liver transplant recipient is only around 50%. Due to the low specificity of EBV load for detecting PTLD, new biomarkers such as CD30 are being sought.
In any case, an elevated EBV DNA in the peripheral blood should alert the physician to perform a physical examination and request imaging. If clinical suspicion is raised, an excisional biopsy should be done. Tissues should be submitted for EBV analysis and determination of CD20 expression, which are critical in determining treatment. If PTLD is confirmed, it is necessary to perform a complete staging, including CT of the neck, chest, abdomen and pelvis, evaluation of the transplanted liver graft, an echocardiogram, and glomerular filtration rate. SPECT-CT is also useful, since it can detect areas infiltrated by lymphoma that had previously gone unnoticed (17).
In our series, PTLD location was extranodal in all cases, the bowel being the most common site. These results do not agree with those obtained in other series where the most common localization were the lymph nodes followed by an intestinal location (18,19). On the other hand, the liver graft is a common site of presentation (up to 49% depending on the series) (19). In our case, although relatively frequent, the percentage is lower (20%).
Early diagnosis of PTLD is essential to initiate treatment and prevent progression to a more aggressive variant. Thus, in 2008 the WHO introduced the PTLD classification, which establishes four categories according to their mode of presentation (Table I).
Early lesions usually occur after the first year post-transplant. They have features of plasmocytic hyperplasia or mononucleosis primary infection (20). Lesions are usually polyclonal and they are more common in EBV seronegative recipients and in young patients. In our series, only one patient had early lesions such as plasmacytic hyperplasia. As in the literature, it occurred in the youngest patient in the series but this case was seropositive for EBV and debuted the first year after transplantation. Polymorphic PTLD, monomorphic PTLD and classic Hodgkin's lymphoma-type PTLD are usually monoclonal and they have a variable time of presentation after liver transplantation. Monomorphic B cell PTLD is the most frequent variant, especially diffuse large B cell lymphomas (21,22). In our cases, 60% of patients had monomorphic B cell lymphoma, of which 50% were diffuse large B cell variant.
Lymphoproliferative syndromes are heterogeneous and EBV plays an important role in its pathogenesis. Eighty-five per cent of PTLD comes from the proliferation of B lymphocytes, and more than 80% of these are associated with EBV (21). Of the 15% that are of T-cell origin, 30% are associated with EBV (21). No monomorphic T cell PTLD was identified in our series; 100% of monomorphic lymphomas corresponded to B-cell lymphomas. These results are comparable with the revised data. However, in our series, only 20% of B cell lymphomas showed EBV seropositivity.
On the other hand, in our series, we observed a high mortality rate among patients who were EBV negative. Specifically, 80% of the deaths were EBV seronegative. The main prognostic factors for PTLD include high-grade lymphomas, EBV negativity on the recipient and an affected liver graft (6). This may explain the high mortality among patients with EBV negativity. As for the two patients who had liver involvement, both patients died and the diagnosis was made post mortem. PTLD located in the liver graft has a fulminant clinical course. Due to its similarity to acute rejection, it is essential to perform a liver biopsy to perform the diagnosis and start early treatment (23).
PTLD treatment
Current treatment for PTLD is based on an ascent therapy. It includes a reduction of immunosuppression, administration of monoclonal antibodies against specific biomarkers of B lymphocytes (anti-CD20) and chemotherapy. Surgery and radiation therapy may be useful when the disease is localized.
It is estimated that reduction of immunosuppression can lead to regression of PTLD in up to 23-50% of cases (17). Immunosuppression should be decreased to the lowest tolerated levels, considering the risk of graft rejection. The liver is a resistant organ to reduction of immunosuppression, with a low rate of graft loss due to rejection. Liver transplantation has a better tolerance to immunosuppression reduction than other solid organ transplants. In our series, in two cases, reducing immunosuppression alone was sufficient to produce complete remission in one patient and stabilization in the other. None of these cases showed signs of acute or chronic rejection after decreasing immunosuppression.
Although there is a tendency to use lower target levels of immunosuppressant in the second half of this study, this does not involves a trend towards a lower number of cases. Fifty percent of the cases were diagnosed in the latter half of the study (Table III).
It is thought that the use of both CsA and FK506 per se increases the incidence of neoplasia with respect to common guidelines of this medication. Although the primary risk factor is the overall degree of immunosuppression, there is evidence to suggest that CsA and FK506 can promote tumor growth in immunosuppressed animals through the release of tumor growth factor beta (24).
When we have a suspicion or confirmation of PTLD, the first step should be to reduce or withdraw immunosuppressive medication if possible, adding drugs with antitumor effect. The use of immunosuppressive regimens that include an mTOR inhibitor such as sirolimus or everolimus, which have an antitumor effect, could improve survival of these patients. However, although its effectiveness is recognized in patients with heart and kidney transplants, its efficacy in liver transplantation has not yet been established, so it is not advised as an initial immunosuppressive (25). In our series, immunosuppressive medication was replaced by an mTOR inhibitor (sirolimus) in three cases. Among them, complete cure of the disease was observed in two cases and stabilization in the remaining case.
New therapies with monoclonal antibodies against lymphocyte specific biomarkers have been a breakthrough in the treatment of lymphoproliferative syndrome. Rituximab therapy (an antibody against the CD20 receptor of B cells) seems to be effective in the management of PTLD cases, with a remission rate of 45-65% (17,26). In our study, 30% of patients received rituximab as first-line treatment, with a complete remission observed in 66% of these cases.
Chemotherapy should be reserved for patients with high-grade lymphomas, disease progression after exhausting other therapeutic options or the occurrence of any organ failure. Generally, chemotherapies are based on anthracycline, which results in longer survival and disease free survival, although its benefits are overshadowed by its toxic effects (23). In our series, we observed the progression of the disease in the two patients who received chemotherapy, resulting in one death.
Conclusion
In summary, patients with suggestive symptoms of PTLD should undergo a liver biopsy and complementary imaging tests that allow early diagnosis. Our first action in PTLD treatment should be to reduce immunosuppression, always assessing the risk of acute liver graft rejection. Patients may also benefit from the replacement of a calcineurin inhibitor for an mTOR inhibitor, specifically sirolimus.
Patient management should be based on histology, stage and location of the tumor. Patients with low-grade B cell lymphoma should be treated with rituximab monotherapy and the replacement of a calcineurin inhibitor for an mTOR inhibitor. In the event of therapy failure, high grade lymphomas or organ failure, chemotherapy should be used.
In conclusion, PTLD is a rare complication after liver transplantation that represents a threat to the patient's life. Due to the high mortality and therapeutic limitations, it is essential to identify patients at risk to obtain an early diagnosis and treatment that can change the outcome of the disease.
References
1. Adam R, Caillez V, Majno P, et al. intrinsic mortality risk in liver transplantation: European Liver Trasplant Registry study. Lancet 2000;356:621-7. DOI: 10.1016/S0140-6736(00)02603-9. [ Links ]
2. Engels EA, Pfeiffer RM, Fraumeni JF Jr, et al. Spectrum of cancer risk among US solid organ transplant recipients. JAMA 2011;306:1891-901. DOI: 10.1001/jama.2011.1592. [ Links ]
3. Bilbao Aguirre I, Lázaro Fernández L, Charco Torra R. Trasplante hepático. En: Parrrilla P, Landa JJ, editores. Cirugía AEC. 2o edición. Spain: Panamerica; 2009. p. 685-704. [ Links ]
4. Taylor AL, Marcus R, Bradley JA. Post-transplant lymphoproliferative disorders (PTLD) after solid organ transplantation. Crit Rev Oncol Hematol 2005;56:155-67. DOI: 10.1016/j.critrevonc.2005.03.015. [ Links ]
5. Opelz G, Dohler B. Lymphomas after solid organ transplantation: A collaborative transplant study report. Am J Transplant 2004;4:222-30. DOI: 10.1046/j.1600-6143.2003.00325.x. [ Links ]
6. Parker A, Bowles K, Bradley JA, et al. Management of post-transplant lymphoproliferative disorder in adult solid organ transplant recipients: BCSH and BTS Guidelines. Br J Haematol 2010;149:693-705. DOI: 10.1111/j.1365-2141.2010.08160.x. [ Links ]
7. Evens AM, David KA, Helenowski I, et al. Multicenter analysis of 80 solid organ transplantation recipients with post-transplantation lymphoproliferative disease: Outcomes and prognostic factors in the modern era. J Clin Oncol 2010;28:1038-46. DOI: 10.1200/JCO.2009.25.4961. [ Links ]
8. Kerkar N, Morotti RA, Madan RP, et al. The changing face of post-transplant lymphoproliferative disease in the era of molecular EBV monitoring. Pediatr Transplant 2010;14:504-11. DOI: 10.1111/j.1399-3046.2009.01258.x. [ Links ]
9. Mukthinuthalapati PK, Gotur R, Ghabril M. Incidence, risk factors and outcomes of de novo malignancies post liver transplantation. World J Hepatol 2016;28:533-44. [ Links ]
10. Kremers WK, Devarbhavi HC, Wiesner RH, et al. Post-transplant lymphoproliferative disorders following liver transplantation: Incidence, risk factors and survival. Am J Transplant 2006;6:1017-24. DOI: 10.1111/j.1600-6143.2006.01294.x. [ Links ]
11. Cockfield SM. Identifying the patient at risk for post-transplant lymphoproliferative disorder. Transpl Infect Dis 2001;3:70-8. DOI: 10.1034/j.1399-3062.2001.003002070.x. [ Links ]
12. Aucejo F, Rofaiel G, Miller C. Who is at risk for post-transplant lymphoproliferative disorders (PTLD) after liver transplantation? J Hepatol 2006;44:19-23. DOI: 10.1016/j.jhep.2005.10.008. [ Links ]
13. Mumtaz K, Faisal N, Márquez M, et al. Post-transplant lymphoproliferative disorder in liver transplant recipients: Characteristics, management and outcome from a single-centre experience with > 1,000 liver transplantations. Can J Gastroenterol Hepatol 2015;29:417-22. DOI: 10.1155/2015/517359. [ Links ]
14. Zimmermann T, Hoppe-Lotichius M, Tripkovic V, et al. Liver transplanted patients with preoperative autoimmune hepatitis and immunological disorders are at increased risk for post-transplant lymphoproliferative disease (PTLD). Eur J Intern Med 2010;21:208-15. DOI: 10.1016/j.ejim.2010.02.009. [ Links ]
15. Kamdara KY, Rooneya CM, Heslopa HE. Post-transplant lymphoproliferative disease following liver transplantation. Curr Opin Organ Transplant 2011;16:274-80. DOI: 10.1097/MOT.0b013e3283465715. [ Links ]
16. McLaughlin K, Wajstaub S, Marotta P, et al. Increased risk for post-transplant lymphoproliferative disease in recipients of liver transplants with hepatitis C. Liver Transpl 2000;6:570-4. DOI: 10.1053/jlts.2000.7578. [ Links ]
17. Bakker NA, Van Imhoff GW, Verschuuren EA, et al. Presentation and early detection of posttransplant lymphoproliferative disorder after solid organ transplantation. Transpl Int 2007;20:207-18. DOI: 10.1111/j.1432-2277.2006.00416.x. [ Links ]
18. Cheuk-lam Lo R, Chan SC, Chan KL, et al. Post-transplant lymphoproliferative disorders in liver transplant recipients: A clinicopathological study. J Clin Pathol 2013;66:392-8. DOI: 10.1136/jclinpath-2012-201139. [ Links ]
19. Izadi M, Fazel M, Saadat SH, et al. Hepatic involvement by lymphoproliferative disorders post liver transplantation: PTLD. Int Survey Hepatol Int 2011;5:759-66. DOI: 10.1007/s12072-011-9271-1. [ Links ]
20. Swerdlow SH, Webber SA, Chadburn A, et al. Classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008. [ Links ]
21. Koch DG, Christiansen L, Lazarchick J, et al. Post-transplantation lymphoproliferative disorder - The great mimic in liver transplantation: Appraisal of the clinicopathologic spectrum and the role of Epstein-Barr virus. Liver Transpl 2007;13:904-12. DOI: 10.1002/lt.21152. [ Links ]
22. Allen U, Alfieri C, Preiksaitis J, et al. Epstein-Barr virus infection in transplant recipients: Summary of a workshop on surveillance, prevention and treatment. Can J Infect Dis 2002;13:89-99. DOI: 10.1155/ 2002/634318. [ Links ]
23. Hoshida Y, Li T, Dong Z, et al. Lymphoproliferative disorders in renal transplant patients in Japan. Int J Cancer 2001;91:869-75. DOI: 10.1002/1097-0215(200002)9999:9999<::AID-IJC1125>3.0.CO;2-N. [ Links ]
24. Hojo M, Morimoto T, Maluccio M, et al. Cyclosporine induce cancer progression by cell-autonomus mechanism. Nature 1999;397:530-4. DOI: 10.1038/17401. [ Links ]
25. Andrassy J, Graeb C, Rentsch M, et al. mTOR inhibition and its effect on cancer in trasplantation. Trasplantation 2005;80:171-4. DOI: 10.1097/01.tp.0000186912.23630.85. [ Links ]
26. Mendizabal M, Marciano S, Dos Santos Schraiber L, et al. Post-transplant lymphoproliferative disorder in adult liver transplant recipients: A South American multicenter experience. Clin Transplant 2013:27:469-77. DOI: 10.1111/ctr.12152. [ Links ]
![]() Correspondence:
Correspondence:
Mercedes Rubio-Manzanares Dorado.
Unit of Hepatobiliary Surgery and Liver Transplantation.
Hospital Universitario Virgen del Rocío.
Av. Manuel Siurot, s/n.
41013 Sevilla, Spain
e-mail: ale.moral@gmail.com
Received: 03-02-2016
Accepted: 08-01-2017
