










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Cirugía Oral y Maxilofacial
versión On-line ISSN 2173-9161versión impresa ISSN 1130-0558
Rev Esp Cirug Oral y Maxilofac vol.29 no.6 Madrid nov./dic. 2007
CASO CLÍNICO
Treatment for fibrous dysplasia when associated with hemophilia C: A case report
Tratamiento de la displasia fibrosa asociada a hemofilia C: a propósito de un caso
T. Creo Martínez1, A. Borrego Luque2, C. Salazar Fernández2, R. del Rosario Regalado1, Y. Marín Lapeira3
1 Médico Residente. Servicio Cirugía Oral y Maxilofacial.
2 Médico Adjunto. Servicio Cirugía Oral y Maxilofacial.
3 Médico Residente. Servicio Radiodiagnóstico.
Hospital Universitario Virgen Macarena, Sevilla, España
ABSTRACT
Fibrous Dysplasia is a benign bone disease that changes normal bone tissue for a proliferation of connective fibrous tissue. It is thought that an alteration of the Gsa gene is the main cause of the disease.
Hemophilia C is a rare inherited blood disease leading to abnormal hemorrhages in affected patients. They have a factor XI deficiency. It is the least frequent of all hemophilias. It is a recessive autosomal disease, affecting both men and women. A 13 year-old patient developed fibrous dysplasia in right upper maxilla. The patient started with pain on chewing hard food. She had vestibular swelling and reddening of the right side of the palate. She had a discrete factor XI deficiency (heterozygotic). She needed special preparation before the lesion could be removed because of her deficiency.
It has been discovered that the mutation of gene Gsa (GNAS1) is the reason for fibrous dysplasia. This gene is in chromosome 20q. The cause of hemophilia C is a factor XI deficiency due to a mutation in the FXI gene in chromosome 4. Perhaps these two rare diseases are related, because both are unusual diseases and both are in the same patient.
Key words: Fibrous dysplasia; Hemophilia C; gene Gsa, Pamidronate; FXI gene.
RESUMEN
La displasia fibrosa es una enfermedad ósea benigna que cambia el tejido óseo normal por una proliferación de tejido conectivo. Se piensa que la alteración del gen Gsa es la principal razón de la enfermedad. La hemofilia C es una enfermedad sanguínea, hereditaria rara, que provoca hemorragias en pacientes afectos. Es autonómica recesiva, por lo que hombres y mujeres pueden estar afectos.
Paciente de 13 años que desarrolla una displasia fibrosa en maxilar superior derecho que empieza con dolor durante la masticación de alimentos duros. Presenta abombamiento de vestíbulo y enrojecimiento de paladar derecho.
Presenta un déficit discreto de factor XI (heterocigoto). Por ello, necesita una preparación especial antes de extirpar la lesión debido a su déficit. Se ha descubierto que la razón de la displasia fibrosa es la mutación del gen Gsa (GNAS1) que está en el cromosoma 20q.
La causa de la hemofilia C es el déficit del factor XI debido a una mutación del gen FXI en el cromosoma 4. Quizás estas dos raras enfermedades tengan una relación, porque ambas se presentan en el mismo paciente.
Palabras clave: Displasia fibrosa; Hemofilia C; Gen Gsa; Pamidronato; Gen FXI.
Introduction
Fibrous dysplasia is a benign bone disorder characterized by the progressive replacement of normal bone tissue by a proliferation of connective fibrous tissue.1Although its etiology has not been totally explained, it has been postulated that it can be due to a disturbance of the Gsa1,2 gene due to a heterozygous mutation of this gene.2 This mutation has been described in monostotic and polyostotic fibrous dysplasia, and in fibrous dysplasia associated with McCune-Albright syndrome.2
Lichtenstein5 was the first to describe the disease in 1938. Fibrous dysplasia can be monostotic or polyostotic. The monostotic form, which appears in the craniofacial region, represents only 10% of cases.4 It tends to appear in the upper jaw2,3 and it can affect adjacent bones such as the zygomatic, sphenoid and occipital bones.2
The disease appears in childhood, usually progressing during puberty and adolescence, and halting in most cases at the end of the adolescence period.6 The disease continues for a certain group of patients, progressing after puberty and causing deformity and/or functional problems.6
The disease manifests in the facial region, either as asymptomatic growth of the affected bone, leading to facial deformity and swelling of soft tissues,7 proptosis, severe headaches, or causing pain in the affected area. It can cause disturbance to the nervous system because of the compression of these structures.
Sarcomatous degeneration can occur, but this is very infrequent, and it is estimated to occur 0.5% of the time.8
Radiologically it is characterized by radiolucid, radiotransparent or radiopaque, sclerotic lesions, depending on the quantity of fibrous component. The typical CAT scan image is a ground glass or eggshell appearance, which corresponds to the healthy bone around the lesion.9
Hemophilia C is a rare hereditary disease of the blood, characterized by a tendency for abnormal hemorrhaging. It is the least frequent of the different forms of hemophilia. It is characterized by a factor XI deficiency, either because of a reduction of the factor or because of the existence of a structurally abnormal factor (CIE-9-MC code). Hemophilia C represents 2-3% of all hemophilias, and it tends to manifest during the neonatal period because of the factor not crossing the placenta, although there may be many asymptomatic newly born infants. The main symptoms are hemarthrosis, hematuria, epistaxis, etc.10
It is a disease that is inherited in the form of an incomplete recessive genetic trait, which is not linked to the X chromosome. This disturbance is found on autosomal chromosomes and the disease therefore manifests in females as well as males.
Case report
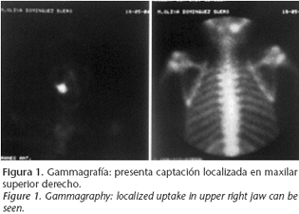
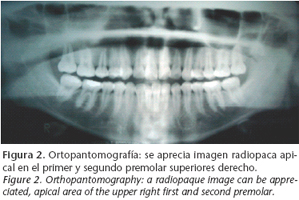
A thirteen-year-old patient was referred to our department in June 2004 as a result of an image appearing on the gammagraphy of her upper right jaw, (Fig. 1) which had been carried out because of lumber pain (Fig. 1). She claimed that her medical history included no disease, and on examination there were no intra- or extraoral symptoms suggestive of any disease. The orthopantomography (Fig. 2) showed a radiopaque image in the apical area of the upper right first and second premolars. The CAT scan carried out for diagnostic confirmation revealed groundglass features compatible with fibrous dysplasia of the upper right jaw (Fig. 3). A biopsy of the affected area provided the definitive diagnosis.

The patient was asymptomatic and she periodically had check-ups. In September 2004 she began to have discomfort on masticating hard food. On examination a bulging of the vestibule and right palate was observed, together with a reddening of the mucosa of the lining. There was no increase in dental mobility. Surgery was suggested with the aim of carrying out complete resection of the lesion.
The preoperative blood tests showed disturbance in the coagulation parameters. After confirmation, a report by a hematologist was requested, which showed a slight deficiency of factor XI (heterozygous) and normochromic normocytic anemia.
The patient was operated in February 2005 under general anesthesia and presurgical preparation was required. The week before the surgical intervention the patient was prepared with tranexamic acid. Twelve hours before the surgery a coagulation study was carried out, which showed that all the values were within the normal range. Instructions were given to transfuse two units of fresh plasma thirty minutes before the surgical act. A right-sided maxillectomy was carried out (Fig. 4), and the tuberosity of the maxilla was removed together with the first premolar and palatine bone. Just the floor of the orbit was left. She evolved favorably and there were no complications in the immediate postoperative period. During the postoperative period, tranexamic acid was maintained for a week together with coagulation tests. The patient was discharged after 10 days, as there were no complications or signs of bleeding.

During the early postoperative period she had pain in the area of the infraorbital nerve and unresolved anemia. In June 2005 the neuralgia disappeared and prosthetic rehabilitation was carried out using a removable partial prosthesis, and there were no signs of recurrence.
In August 2005 she had recurrence in the area of the right malar, despite being asymptomatic. A month later she had localized pain in this area. The option put forward for treating her was diphosphonates for pain relief.
Discussion
Fibrous dysplasia is a rare benign disease that can affect a single bone (monostotic type), various bones (polyostotic type), or it can be accompanied by another systemic pathology, including abnormal pigmentation of the skin, premature sexual development and hyperthyroidism (McCune Albright syndrome). It can appear on its own, associated with other diseases such as Pyles disease or craniometaphyseal dysplasia.6
It has recently been discovered that the mutation of the gene Gs á(GNAS1) found in chromosome 20q is the cause of fibrous dysplasia.11 This mutation occurs during embryonic development or during postnatal life. The mutation of gene Gsa produces an increase in adenylate cyclase, which produces an increase in intracellular AMPc. The high concentration of intracellular AMPc generates an increase in the inappropriate proliferation and differentiation of the cells after their mutation, which causes the formation of an immature and disorganized fibrous matrix that generates the fibrous tissue of the dysplasia.2
Hemophilia C is a rare disease as a result of a factor XI coagulation deficiency, which has a part in the early phase of the intrinsic coagulation pathway. Most of the factor XI deficits occur in the Jewish Ashkenazi population.12 It is known that the disease is a mutation that occurs in the FXI gene that is in chromosome 4.10 In the FXI gene there are three specific mutations that divide the disease into three types depending on the mutation area, which are type I, type II and type III.13
Our patient had monostotic fibrous dysplasia of the upper jaw and heterozygotic hemophilia C, two unusual diseases, which in this case appeared together in the same person. In addition these were two diseases that, according to the latest studies, arise as a result of genetic mutation in autosomal genes. As a result of this, we requested kariotyping of the patient in order to find out if she had any type of related genetic condition (Fig. 5). The result of the kariotyping was that all the metaphases analyzed had 46 chromosomes. Following GTG banding techniques, no anomalies were found in the metaphases studied. Although the karyotype was normal, the possibility could not be ruled out that there were some chromosomal mosaicism, microdeletions, microduplications, and/or small structural rearrangements.

In the literature, the connection between fibrous dysplasia and other diseases such as Pyles disease is reflected, although the connection between both diseases has yet to be established.6 As a result we may find a second disease appearing together with fibrous dysplasia.
Treatment for fibrous dysplasia is very variable, depending on how the patient is affected and the symptoms. Thus, the treatment can vary from not doing anything and monitoring the patient periodically if there is no symptomatology, to complete resection of the affected bone, or just simple curettage. The choice of treatment depends on the area affected and the reconstruction capacity of the resected tissue. Total resection is indicated if the patient has symptoms that include pain, facial deformity, or invasion of important structures, such as the orbit with compression of the eyeball or the optic nerve.3,14 There are other treatment possibilities that appear to be giving encouraging results such as the use of intravenous pamidronate.15,16 The use of antiresorptive agents such as bisphosphonates in the treatment of fibrous dysplasia is justified because of the histological findings of fibrous dysplasia of a high number of osteoclasts, and the evidence of bone resorption in fibrous dysplasia lesions. Thus, increased bone resorption appears to play an important role in the establishment and growth of some dysplasia lesions.16
Our patient was initially offered surgical treatment for the lesion in addition to treatment with pamidronate, but she demanded definitive surgical treatment herself, (due to the lesions relatively small size and that it could be approached intraorally). During the presurgical preparation, hemophilia C was detected, and additional measures were taken, and there was therefore increased surgical risk.
During the intervention an attempt was made at carrying out definitive treatment and the affected bone was resected completely, despite the limitations of using an intraoral approach, as the patient had refused an extraoral approach. It seemed that by using an intraoral approach, the disease could be resected completely, but months after the intervention the patient suffered a recurrence, with intense pain in the affected area. Surgical treatment was proposed once again and, bearing in mind the high risks with this patient because of concomitant disease and the pre- and postoperative preparation required, together with the risk involved in fresh plasma transfusion, intravenous pamidronate therapy was also proposed. Finally, pamidronate therapy was chosen, which in this case was absolutely indicated, as the fibrous dysplasia was painful. 1 mg/kg of IV pamidronate was administered for three days every 4-6 months. After the administration of pamidronate the pain in the jaw decreased and the patient is to date free of symptoms.
One of the problems of using this therapy with children and adolescents, is that it interferes with their growth.15 Long term studies have been carried out that show that diphosphonates do not seriously affects growth,16 and it would appear that the only secondary effect that was registered during perfusion was an increase in body temperature in some patients of 38-40ºC, and which was solved without any problem by using NSAIDs.15 Another side effect registered in patients with renal involvement (some Albright syndrome patients) is osteomalatia that decreases on suspending treatment, and which is inevitable in these patients if diphosphonates are used in conjunction with phosphorus taken orally.16 Thus using diphosphonates can be considered safe treatment.
Therefore, given the favorable studies with this drug and its safety, together with the greater risks that more aggressive surgical treatment implied for this patient, intravenous pamidronate treatment was decided on.
Although surgical treatment of fibrous dysplasia is a good treatment option, it would seem that for our patient this was not the most adequate option, which demonstrates once again that in fibrous dysplasia there is no standard treatment. Each case should be evaluated individually due to the associated diseases and complications that may arise, and one therapeutic option should be selected over another depending on the patient.
![]() Correspondence:
Correspondence:
Teresa Creo Martínez
Avda. Dr. Fedriani 19, 2º-2º
41009 Sevilla, España
Email: tesicreo@gmail.com
Receceived: 12.04.06
Accepted: 18.12.06
References
1. Concha G, Jofré S. Displasia fibrosa en el territorio maxilofacial. Definición e imagen en radiología convencional y TC. Anuario Sociedad de Radiología Oral y Maxilofacial de chile 2003;6-1. [ Links ]
2. Perdigao PF, Pimenta FjGS, Castro WH, De Marco L, Gomez RS. Investigation of the Gsa gen in the diagnostic of fibrous displasia. Int J Oral Maxillofacial surgery 2004;33:498-501. [ Links ]
3. Zenn MR, Zuniga J. Treatment of fibrous dysplasia of the mandible with radical excision and immediate reconstruction: a case report. J Craniofac Surg 2001;12- 3. [ Links ]
4. Gillman G, Bryson PC, Rao UNM. Radiology quitz case 1: Isolated fibrous dysplasia of the sphenoidal sinus. Arch Otolaryngol Head Neck Surgery 2004;130. [ Links ]
5. Lichtenstein L. Polyostotic fibrous dysplasia. Arch Surg 1938;36:874-98. [ Links ]
6. Ozek C, Gundogan H, Bilkay U, Tokat C, Gurler T, Songur E. Craneomaxillofacial fibrous dysplasia. J Craniofac Surg 2002;13-3. [ Links ]
7. Crawford LB. An unusual case of fibrous dysplasia of the maxillary sinus. Am J Orthodontics Dentofac Orthoped 2003;124-6. [ Links ]
8. Michael CP, Lee AG, Patrinely JR, Stal S, Blacklock JB. Visual loss associated with fibrous dysplasia of the anterior skull base. J Neurosurg 2000;92: 350-4. [ Links ]
9. Tokano H, Sugimoto T, Noguchi Y, Kitamura K. Sequential computed tomography images demonstrating characteristic changes in fibrous dysplasia. J Laryngol Otol 2001;115:757-9. [ Links ]
10. Bolton-Maggs PHB. Deficiencia del factor XI y su manejo. Federación mundial de hemofilia. Tratamiento de la Hemofilia 2004;16. [ Links ]
11. Levine MA, Modi WS, O´Brien SJ. Mapping of the gene encoding the alpha subunit of the stimulatory 6 protein of adenylyl cyclase (GNAS1) to 20q13.2-q13.3 in human by in situ hybridization. Genomics 1991;11:478-9. [ Links ]
12. Meijers JCM, Davie EW, Chung DW. Expression of human blood coagulation factor XI: characterization of the defect in factor XI type III deficiency. Blood 1992;79:1435-40. [ Links ]
13. Hancock JF, Wieland K, Pugh RE, Martinowitz U, Schulman S, Kakkar VV, Kernoff PBA, Cooper DN. A molecular genectic study of factor XI deficiency. Blood 1991;77:1942-8. [ Links ]
14. Keijser LCM, Van Tienen TG, Bart Schreuder HW, Lemmens JAM, Pruszczynski M, Veth RPH. Fibrous dysplasia of bone. Management and outcome of 20 cases. J Surg Oncol 2001;76:156-66. [ Links ]
15. Kos M, Luczak K, Godzinski J, Klempous J. Treatment of momostotic fibrous dysplasia with pamidronato. J Craniomaxillofac Surg 2004;32:10-5. [ Links ]
16. Chapurlat RD, Huguensy P, Delmas PD, Meunier PJ. Treatment of fibrous dysplasia of bone with intravenous pamidronate: long term effectiveness and evaluation and evaluation of predictors of response to treatment. Bone 2004; 35:235-42. [ Links ]

