










 Serviços customizados
Serviços customizados
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkINTRODUCTION
Pseudotumoral calcinosis is a benign and rare entity. The term has indistinctly been used to describe any kind of calcification around a joint, although it was historically designated to describe a hereditary condition. Multiple diseases and systemic conditions have later been related in non-hereditary cases, especially chronic renal failure.
Classic tumoral calcinosis lesions present as lobular calcified masses in the periarticular soft tissue. Frequently found in hips, elbows and shoulders, being extremely rare in the head and neck region.
CT and MRI are necessary for diagnosis. However, definitive diagnosis is given by the pathological results. Differential diagnosis includes calcific myonecrosis, gout, osteomiosarcoma, myosistis ossificans, chondromas, chondrosarcomas and osteochondromatosis among many.
The most effective treatment is surgical resection, but recurrences may happen. Some medical therapies, such as phosphate depletion, have proved variable success. In this article we present the case of a 56-year-old woman who was diagnosed with pseudotumoral calcinosis of the anterior cranial base.
CASE REPORT
A 56-year-old woman was referred to our department in 2018 with a tumor located in the right masticatory space. She suffered from paresthesia in the right side of the face, associated to fluctuating neuropathic pain.
In July 2018, a CT was done, describing a mass located in the right masticatory space in the lateral pterygoid muscle, measuring 27 x 20 x 23,5 mm. It showed a chondroid-like matrix with calcifications. It extended through the medial pterygoid plates and produced smooth bone erosion of the sphenoid, extending as well through the temporal fossa and in proximity to the oval foramen (Figure 1).
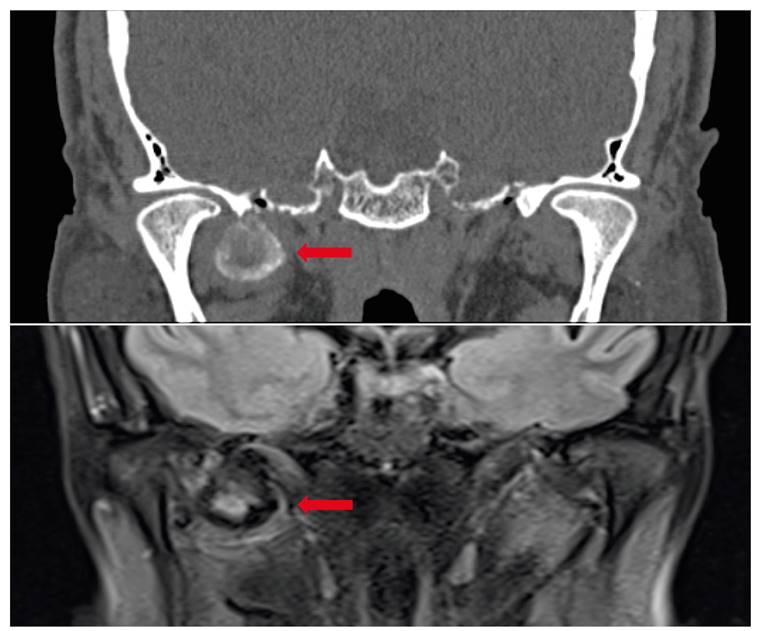
The MRI demonstrated a lobulated mass hypointense in T3, suggesting an osseous component or calcifications, and a hyperintense center in T2 with a pseudocapsule located in the lateral pterygoid muscle, in close proximity to the carotid artery and oval foramen. The greater sphenoid wing showed mild erosion. Cerebral compromise was discarded. No pathological nodes were observed.
Posteriorly, a CT-guided FNA was performed, suggesting a chondrosarcoma.
Surgical excision was performed, assisted by intraoperative navigation (Figure 2). An hemicoronal approach was performed. An in-house patient-specific surgical guide was previously printed for the orbitozygomatic osteotomy (Figure 3). Through careful dissection the tumor was identified. Intraoperative biopsies discarded malignancy. The mass was excised (Figure 4) and the bone was repositioned and fixed with 3 plates. Closure in layers was performed and a drainage was left in place for two days. No immediate postoperative complications occurred.
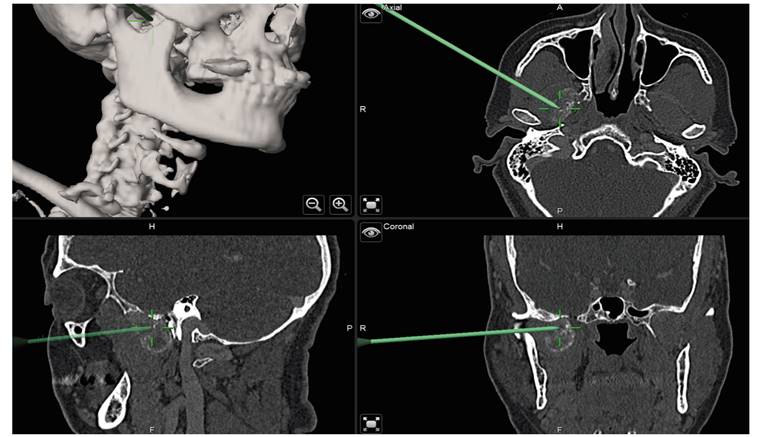

Figure 3. From left to right: hemicoronal approach and surgical guide in place; surgical detail, white arrow shows the zygomatic arch and the yellow arrow shows the tumor.

No facial palsy or visual alterations were observed in the postoperative period. She was referred to rehabilitation due to limited mouth opening because of fibrosis and myofascial pain. She also received botox injections in the temporalis and masseter muscles due to myofascial pain.
Macroscopic analysis described a white chalky calcified lobulated mass measuring 4,5 x 4 x 1cm. Microscopically, a fibrohistiocytic proliferation and foreign body granulomatous reaction was found surrounding acellular areas (Figure 5). Hydroxiapatite distrophic calcifications were identified.
Connective tissue presented with broad dystrophic calcifications and deposit of calcium hydroxiapatite. The definitive diagnosis was pseudotumoral calcinosis.

Figure 5. Fibrohistiocytic proliferation and foreign body granulomatous reaction was found surrounding acellular areas. Hydroxiapatite distrophic calcifications were also found.
Diabetes, gout, chronic renal failure and hypoparathyroidism were ruled out. Postoperative imaging 6 months later (Figure 4) showed no signs of relapse.
DISCUSSION
Tumoral calcinosis is an unusual disease of unclear etiology and pathogenesis1. First described by Duret in 1989, this pathology affects both sex indifferently and some authors report a higher rate in young and black race patients. Other terms used in the literature are uremic tumoral calcinosis, secondary tumoral calcinosis, pseudotumor calcinosis, nonfamilial tumoral calcinosis and tumoral calcification.
Generally, it is associated to metabolic disorders where an increase in the calcium x phosphate relation is found. Pseudotumoral calcinosis occurs in three clinical settings: as a complication of renal dialysis in patients with cronic renal failure, which is the most frequent cause; in patients with heritable abnormality of vitamin D metabolism; and sporadically in patients with degenerated or inflamed tissue without metabolic abnormalities2. In these latter cases where no metabolic or genetic cause can be identified, repetitive trauma, local dystrophia, tissue necrosis or fibrous alterations can apparently be involved1. It has also been identified in some inflammatory connective tissue disorders such as systemic lupus, scleroderma or the CREST syndrome.
It is characterized by periarticular soft tissue dystrophic calcifications with a radiodense appearance in CT imaging. It is usually found occurring in periarticular regions, such as the hips, elbows and shoulders. However, few cases have been reported in the literature about tumoral calcinosis ocurring in the head and neck. A few cases have reported pseudotumoral calcinosis in the TMJ1.
Clinically, pseudotumoral calcinosis can be asymptomatic in most cases in the initial stages of the disease but, as it progresses and grows, it may cause pain and neurological symptoms due to nerve compression or pain. When occurring in the periarticular tissue, reduced joint mobility might be found, causing disability and pain. Differential diagnosis is wide, and includes osteosarcoma, leiomyosarcoma, chondrosarcoma and giant cell tumors, among others.
CT and MRI are generally recommended. It is generally identified as a homogeneous well delimited and lobulated calcified mass with fibrous septae which can demonstrate fluid calcium levels on CT and soft tissue calcifications. In the MRI it is presented as a hypointense in T1-T2 weighted sequences.
The definitive diagnosis is dictated by the pathologist. Macroscopically, these lesions are usually cystic in appearance and contain white-to-pale yellow chalky material3. Decalcification is generally needed to study the specimen. Microscopically, pseudotumoral calcinosis presents as a chondromyxoid matrix with a nodular pattern surrounded by acellular areas. The deposition of hydroxyapatite and calcium phosphate crystals surrounded by a granulomatous foreign body reaction4 with giant multinucleated and epithelioid cells is characteristic of this pathology.
Surgical excision of the lesions is the only definitive treatment. Incomplete excision can cause relapses. Indications for surgical treatment include pain, restricted movement, ulceration or neurological symptoms. Some authors have reported good results with intravenous and intralesional sodium thiosulfate for ectopic calcifications related to chronic renal failure and non-uremic conditions5,6)
We report the case of a pseudotumoral calcinosis of the anterior cranial base, which is extremely rare. In this clinical case, the patient presented with a slow growing tumor in the anterior right cranial base region. However, the initial diagnosis was chondrosarcoma, and surgery was therefore indicated.
A right hemicoronal approach was performed, using in-house 3D printed surgical guides and intraoperative navigation to assist in the location of the mass, given the complexity of the region. Postoperative histological analysis confirmed a pseudotumoral calcinosis lesion. The patient did not have any renal or metabolic disease, neither had she any history of phosphate metabolism familiar disorder. This case would be included in the non-uremic and non-familiar pseudotumoral calcinosis. Follow up imaging showed good results and complete resection of the tumor.
In conclusion, pseudotumoral calcinosis is a rare entity which may be related to metabolic or renal diseases, hereditary or may be idiopathic, as in the clinical case reported in this article. Radiologically it simulates a calcifying tumor and the initial diagnosis might be difficult, especially in the head and neck region, where this pseudotumoral calcinosis is extremely rare. Complete surgical resection is indicated. In cases where resection might be difficult, such as the anterior cranial base, intraoperative navigation is useful.

