










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNeurocirugía
versión impresa ISSN 1130-1473
Neurocirugía vol.17 no.1 feb. 2006
Complete surgical resection of high-grade astroblastoma with long time survival: case report and review of the literature
Astroblastoma de alto grado con supervivencia prolongada tras resección completa: descripción de un caso y revisión de la literatura
P. Miranda; R.D. Lobato; A. Cabello*; P.A. Gómez y A. Martínez de Aragón**
Servicios de Neurocirugía, Neuropatología* y Neurorradiología**. Hospital 12 de Octubre. Madrid.
SUMMARY
Astroblastoma is a rare glial neoplasm of unknown origin and uncertain prognosis. It usually presents in young adults as a well circumscribed hemispheric mass, often associated with a cystic component. The histological features of astroblastoma are the presence of typical astroblastic perivascular pseudorosettes and perivascular hyalinization. Two different subtypes of astroblastoma have been defined based upon histological characteristics. Prognosis, however, sometimes is in contradiction with the pathological appearance and seems to be more closely related to the grade of surgical resection. We present a new case of a patient with a high-grade astroblastoma with a long survival time, in whom complete surgical resection was confirmed by an early postoperative MRI.
Key words: Astroblastoma. Glial tumor. Treatment. Prognosis.
RESUMEN
El astroblastoma es un tumor glial infrecuente de origen desconocido y pronóstico incierto. Habitualmente se presenta en adultos como una masa hemisférica bien definida, presentando a menudo un componente quístico. Las características histopatológicas de este tumor incluyen la presencia de pseudorrosetas perivasculares astroblásticas típicas y la hialinización perivascular. Se han distinguido dos tipos de astroblastoma en relación a la malignidad histológica. Sin embargo, el pronóstico en ocasiones no guarda relación directa con el tipo histológico y parece más dependiente del grado de resección quirúrgica. Presentamos el caso de una paciente con un astroblastoma de alto grado con supervivencia prolongada, en el cual la resección quirúrgica completa se demostró mediante resonancia magnética postoperatoria precoz.
Palabras clave: Astroblastoma. Tumor glial. Tratamiento. Pronóstico.
Introduction
Astroblastomas are glial neoplasms of unknown origin characterized by a typical perivascular pattern of GFAPpositive astrocytic cells with broad non-tapering processes radiating towards a central blood vessel. Since a similar histological pattern may occur in low- and high-grade astrocytomas as well as in glioblastomas, the term astroblastoma should be restricted to those rare tumors in which it prevails throughout a well-demarcated lesion that does not contain foci of conventional or infiltrative astrocytoma or ependymoma. Prognosis is still uncertain. Complete surgical resection has been sometimes associated with long survival times even in cases showing histological features of malignancy.
Case report
A 42-year-old woman presented with a 3-month history of headache and a recent episode of a complex partial seizure with secondary generalization. Physical and neurological examination were both normal.
Cranial CT showed a left frontal cortico-subcortical lesion, isodense to the brain parenchyma, with heterogeneous enhancement after intravenous contrast administration. Cranial MRI also revealed heterogeneous areas inside the lesion which showed welldefined borders. There was mild edema of the surrounding brain and little mass effect on the ipsilateral ventricle.
The preoperative diagnosis was high grade glioma. At surgery, a well defined cortico-subcortical lesion with small cysts in its deepest part was found. Apparently, it was completely resected and an early cranial MRI, performed within 24 hours of operation, confirmed the absence of tumoral rests.
Histopathological examination showed an extense perivascular pseudorosette pattern of astrocytic cells with broad processes radiating towards central vessels; small foci of hyalinization were frequent. In some areas cellularity was increased with pleomorphism, high mitotic rate, small necrotic areas with perinecrotic palisading and hypertrophy and hyperplasia of vascular endothelia. Tumoral cells were GFAP, vimentin and s-100 positive, preferentially in the perivascular processes.
The postoperative course was uneventful. The patient underwent adjuvant radiation therapy. One month after surgery the patient had returned to her previous job as a teacher and eighteen months later she remains asymptomatic and both MRI and cranial SPECT show no recurrence.
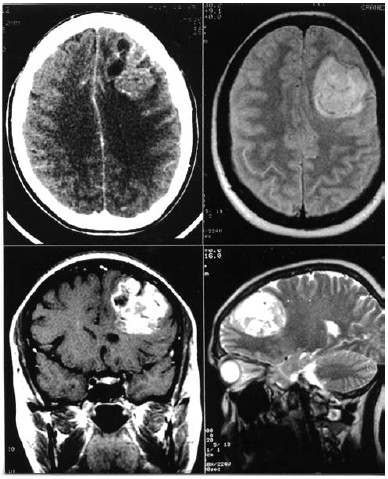
Figure 1. Upper: Left: Preoperative enhanced CT scan
showing a left frontal cortico-subcortical lesion, with
heterogeneous enhancement. Right: Axial slice showing
the well-defined lesion. Lower: Left. Coronal gadoliniumenhanced
T1-eighted MRI demonstrates heterogeneous enhancement
of the lesion and little focal mass effect on ipsilateral
ventricle. Right: Sagittal T2-weighted MR1 shows the
existence of little cysts inside the lesion,
without edema of the surrounding brain.

Figure 2. Photomicrographs showing a perivascular pseudorosette
pattern of GFAP-positive cells with broad, non-tappering
processes radiating towards a central blood vessel.
Tumoral cells display pleomorphic nuclei and cell bodies,
with signs of atypia. Upper left, hematoxilin/eosin x200;
upper right, hematoxilin/eosin x400; Lower left
and right: immunostaining for GFAP x400.

Figure 3. Gadolinium-enhanced TI-weighted MRl performed
12 months after surgery shows no tumoral recurrence. C
ontrast enhancement was observed as a postoperative
change and washed out in successive controls.
Discussion
Astroblastoma is a rare type of glial tumor that was firstly described by Bailey and Buey in 19301. Although its existence has been questioned by some investigators21, it is now recognized as a distinct clinicopathological entity4, probably representing less than 0.5% of gliomas17. It usually appears in young-adult patients, although congenital cases have also been described13. No sex preference has been observed15.
Clinical symptoms at presentation depend on localization, size and mass effect of the neoplasm. Headache and seizures are often present. The cerebral hemispheres are most commonly affected, but astroblastoma may also develop in other places of the central nervous system, such as the corpus callosum, brainstem or cauda equina7.
Radiologically, astroblastoma usually appears as an hemispheric, well circumscribed, often cystic mass, located peripherally near or at the surface of the brain. Thus, differential diagnosis with extraaxial neoplasms is sometimes necessary2,10. Astroblastoma tends to be isodense to brain parenchyma on the plain CT scan.
Calcifications may be seen. The solid component is contrast enhancing. On MRI, astroblastoma appears hypointense in T1-weighted sequences and hyperintense on both proton-density and T2- weighted sequences. Brain edema, if present, is mild. MR spectroscopy findings documented are consistent with those observed in other brain neoplasms and are particularly similar to those of astrocytoma2,10.
According to the WHO, astroblastoma belongs to the category of neuroepithelial tumors of unknown origin8. Rubinstein and Herman14 observed a close ultraestructural and immunohistochemical similarity between astroblastoma cells in culture and the tanycyte, which is a cell integral to the ependyma of submammalian species with transitional features between the astrocyte and the ependymal cell. It has been suggested that a similar cell may also occur transiently during normal human embryogenesis and that astroblastoma may derive from abnormally persisting examples of such embryonal precursor cells. Therefore, despite its designation, astroblastomas are not considered to be embryonal neoplasms, but rather of astrocytic lineage if their characteristics are taken into account.
Concerning the cytological architecture of astroblastoma, Mierau et al12 demonstrated two morphologically distinct cell types displaying an unususal organizational relationship: more primitive cells appeared nesting within the cytoplasm of the differentiated cells. The authors hypothesized that the more differentiated cells served as "nurse" cells for the maturing population.
Histologically, astroblastoma is characterized by a typical perivascular pseudorosette pattern of GFAP-positive cells with broad, non-tapering processes radiating towards a central blood vessel. A component of perivascular hyaline is usually present. Focal astroblastic features are nonspecific and can be seen in low and high grade astrocytic tumours. As a consequence, the term astroblastoma should be reserved for those rare tumours in which the pattern prevails throughout a well demarcated lesion. Interestingly, a case of astroblastoma of the pure type has also been reported19.
In a pathological study of 23 astroblastomas with a postoperative follow-up in 13 patients, Bonnin and Rubinstein3 observed two distinct histological types: 1) a lowgrade type, in which a better differentiated pattern was apparent, and a favourable postoperative prognosis may be expected in a fairly large proportion of patients; and 2) a high-grade type, showing more anaplastic features and in which the length of postoperative survival was usually shorter . Focal or multifocal regions of high cellularity, anaplastic nuclear features, elevated mitotic indices (>5 per 10 HPF), vascular proliferation and necrosis with pseudopalisading correspond to this high-grade group4.
Immunohistochemically astroblastomas are immunoreactive for GFAP, S-100 protein and vimentin5,6,11. The majority also display at least a focal cytoplasmic immunoreactivity for EMA. We have previously reported a case of low-grade astroblastoma in which due to the scarce positivity with GFAP, in contrast with the abundance of intermediate filaments in electron microscopy, along with immunohistochemical and ultraestructural findings, lead us to conclude that the filaments seen in tumor cells were mainly vimentin filaments5.
Recently, molecular analysis has been applied to the study of chromosomal abnormalities in astroblastoma. Brat et al4, using comparative genomic hybridization, detected chromosomal abnormalities in every case studied, the gain of 20q being the most frequent. Although the spectrum of chromosomal alterations indicated that astroblastoma may have its own genetic profile, none of the detected anomalies were specific for malignancy. Shuangshoti et al16 using mycrosatellite markers, showed loss of heterozygosity (LOH) at the DI 9S412 locus on the long arm of chromosome19. This observation was compatible with the loss of a tumour suppressor gene in this region. This antioncogen is supposed to play a role in the tumoral genesis.
The prognosis of astroblastoma remains to be determined. In the abscence of sufficient clinicopathological data, it has been decided not to establish a WHO grade at the present moment11. Low grade astroblastomas seem to have a better prognosis than the high grade ones3. Occasionally, however, in the presence of histological features strongly indicative of malignancy, the clinical follow-up may be in contradiction with the microscopical appearances. This is in contrast to astrocytomas, in which there is extensive concordant evidence that clinical behavior is closely correlated with histopathological features. Long term survival likely depends on numerous factors including tumor location, extent of resection and response to adjuvant therapy3,18. In our case, complete surgical resection has probably played a major role in the long tumor-free survival of the patient. However, and given the histological malignancy of the neoplasm, postoperative adjuvant therapy was associated in this patient. Although astroblastoma appears to be radiosensitive, no definite results concerning chemotherapy are available3.
The optimal management of astroblastoma remains to be established. As mentioned above, radical removal is important and given its peripheral location and well circumscribed aspect, it seems a goal easier to achieve than in other gliomas. In low-grade astroblastomas radical surgery may be the only necessary treatment, although cases of early recurrence have been documented3,5. In such instances, re-operation and adjuvant radiotherapy are the better options though in cases of high-grade astroblastomas a complete resection may be also curative. The lack of enough information about the natural history favours the use of adjuvant radiotherapy from the beginning.
Early MRI postoperative MRI is helpful in demonstrating the absence of residual tumor after surgery that could be otherwise misinterpreted as tumor recurrence in ulterior studies. Therefore it seems very useful in helping to determine the influence of radical resection in the natural history, prognosis and best management of patients with astroblastoma. However, further studies are still necessary to define the real behavior of astroblastoma and determine its optimal management.
References
1. Bailey, P., Bucy, P.C.: Astroblastomas of the brain. Acta Psychiatr Neurol 1930 ; 5: 439-461. [ Links ]
2. Baka, J., Patel, S.C., Roebuck, J.R., Hearshen, D.O.: Predominantly extra-axial astroblastoma: imaging and proton MR speetroseopy features. AJNR 1999; 14: 946-950. [ Links ]
3. Bonnin, J.M., Rubinstein, L.J.: Astroblastomas: a pathological study of 23 tumors, with a postoperative follow-up in 13 patients. Neurosurgery 1989; 25 :6-13. [ Links ]
4. Brat, D.J., Hirose, Y., Cohen, K.J., Feuerstein, B.G., Burger, P.C.: Astroblastoma: clinicopathological features and chromosomal abnormalities defined by comparative genomic hybridization. Brain Pathol 2000; 10: 342- 52. [ Links ]
5. Cabello, A., Madero, S., Castresana, A., Lobato, R.D.: Astroblastoma: electron microscopy and immunohistochemical findings: case report. Surg Neurol 1991; 35:116-121. [ Links ]
6. Hoag, G., Sima, A.A., Rozdilsky, B.: Astroblastoma revisited: a report of three cases.Acta Neuropathol (Berl) 1986; 70: 10-16. [ Links ]
7. Husain, A.N., Leestma, J.E.: Cerebral astroblastoma: immunohistochemical and ultrastructural features. Case report. J Neurosurg 1986; 64: 657-661. [ Links ]
8. Kleihues, P., Burger, P.C., Scheithauer, B.W.: Histological typing of tumors of the central nervous sytem. WHO international classification of tumors. 2nd ed. Berlin: Heidelberg, 1993. [ Links ]
9. Kubota, T., Hirano, A., Sato, K., Yamamoto, S.: The fine structure of astroblastoma. Cancer 1985; 55: 745-750. [ Links ]
10. Kugel, H., Heindel, W., Ernestus, R., Bunke, J., du Mesnil, R., Friedman, G.: Human brain tumors: spectral patterns detected with localized H-1 MR spectroscopy. Radiology 1992; 183: 701-709. [ Links ]
11. Lantos, P.L., Rosenblum, M.K.: Astroblastoma. In Kleihues, P., Cavenee, W.K., eds. Pathology and genetics of tumours of the nervous system. Lyon: IARC Press, 2000: 88- 89. [ Links ]
12. Mierau, G.W., Tyson, R.W., McGavran, L., Parker, N.B., Partington, M.D.: Astroblastoma: Ultraestructural observations on a case of high-grade type. Ultraestruct Pathol 1999; 23: 325-332. [ Links ]
13. Pizer, B.L., Moss, T., Oakhill, A., Webb, D., Coakham, H.B.: Congenital astroblastoma: an immunohistochemicaa study. J Neurosurg 1995; 83: 550-555. [ Links ]
14. Rubinstein, L.J., Herman, M.M.: The astroblastoma and its possible cytogenic relationship to the tanycyte. An electron microscopic, immunohistochemical, tissue-and organ-culture study. Acta Neuropathol (Berl) 1989; 78: 472-483. [ Links ]
15. Russell, D.S., Rubinstein, L.J.: Pathology of the Tumours of the Nervous System. 5th ed. London: Edward Amold, 1989; 161-169. [ Links ]
16. Shuangshoti, S., Mitphraphan, W., Kanivisetsri, S., Griffiths, L., Navalitloha, Y., Porthanakasem, W., Mutirangura, A.: Astroblastoma: report of a case with microsatellite analysis. Neuropathol 2000; 20: 228-232. [ Links ]
17. Sharenberg, K., Liss, L.: Neuroectodermal Tumors of the Central and Peripheral Nervous System. Baltimore: Williams & Wilkins, 1969: 17-29. [ Links ]
18. Thiessen, B., Finlay, J., Kulkarni, R., Rosenblum, M.K.: Astroblastoma: does histology predio clinical behavior? J Neuro-Oncol 1998; 40: 59-65. [ Links ]
19. Yamashita, J., Handa, H., Yamagami, T., Haebara, H.: Astroblastoma of pure type. Surg Neurol 1985; 24: 218-222. [ Links ]
20. Yunten, N., Ersahin, Y., Demirtas, E., Yalman, O., Sener, R.N.: Cerebral astroblastoma resembling an extra-axial neoplasm. J Neuroradiol 1996; 23: 38-40. [ Links ]
21. Zülch, K.J.: Brain tumors. Their Biology and Pathology. 2nd ed. New York: Springer-Verlag 1965: 24, 160. [ Links ]
![]() Correspondencia postal:
Correspondencia postal:
P. Miranda.
Servicio de Neurocirugía.
Hospital 12 de Octubre.
Avenida de Córdoba s/n.
28041. Madrid.
Recibido: 10-10-04.
Aceptado: 18-01-05
Abreviaturas: CT: Computed tomography. GFAB: Gliofibrilar acid protein. MRI: Magnetic Resonance Imaging.

