










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkNeurocirugía
Print version ISSN 1130-1473
Neurocirugía vol.18 n.4 Aug. 2007
Análisis de la presencia de mutaciones por pérdida de heterocigocidad de 1p/19q en tumores cerebrales de estirpe glial
Allelic loss at 1p/19q analysis in brain tumors of glial lineage
J.L. Gil-Salú; A. Nieto*; J.F. Rodríguez-Gutiérrez* y J. Almarcha
Servicios de Neurocirugía y de Inmunología*. Hospital Universitario Puerta del Mar. Cádiz. España.
Dirección para correspondencia
RESUMEN
Objetivos. Analizar en tumores cerebrales, fundamentalmente de estirpe neuro-epitelial, la existencia de mutaciones en los cromosomas 1p y 19q por la técnica de análisis de la pérdida de heterocigocidad (LOH). Un primer objetivo implícito fue poner a punto la técnica del análisis.
Método. Hemos investigado la existencia de mutaciones en 3 alelos seleccionados del cromosoma 1p y en 2 alelos del 19q de distintos tumores cerebrales de estirpe glial intervenidos de manera consecutiva en nuestro Centro desde Octubre de 2004 a Marzo de 2006. La metodología empleada ha sido la detección en ADN tumoral de tejido en fresco y en sangre del paciente del marcaje por PCR de amplificados y electroforesis analizando la pérdida de heterocigocidad de microsatélites, repeticiones de dinucleótidos, situados en D1S508, D1S2734, D1S199, D19S412 y D19S219.
Resultados. Hemos incluido en esta primera fase de estudio un total de 45 muestras de pacientes intervenidos de tumores cerebrales supratentoriales de estirpe neuroepitelial y que incluyen: 29 glioblastomas, 1 gliosarcoma, 7 astrocitomas grado II, 1 oligoastrocitoma, 3 oligodendrogliomas, 1 oligodendroglioma anaplásico, 1 xantoastrocitoma, 1 tumor neuroepitelial disembrioplásico y 1 astrocitoma pilocítico. La presencia de mutación la hemos considerado cuando el índice alélico (T1/T2)/(N1/N2) era inferior a 0.8. Por estirpe histológica destaca la presencia de mutación en un 80% de tumores oligodendrogliales, 14% de glioblastomas y 14% de astrocitomas fibrilares grado II.
Conclusiones. La técnica de análisis de LOH en 1p/19q es factible de realizar en centros que dispongan de técnicas de estudios genético-moleculares, con un alto índice de fiabilidad. De su resultado se desprende qué pacientes se pueden beneficiar del tratamiento con alquilantes añadiendo, a la terapia quirúrgica y/o radioterápica en uso hasta la fecha, una posibilidad de tratamiento con alto porcentaje de respuestas.
Palabras clave: Tumores cerebrales. Gliomas. Mutación 1p/19q. Pérdida de heterocigocidad. Alquilantes.
SUMMARY
Background. To analyze in cerebral tumors of neuroepithelial tissue 1p/19q codeletions by study of loss of heterozygosity (LOH). A first implied objective was to get ready this molecular thecnique.
Methods. We aimed to determine several deletions mapping 1p and 19q chromosomes, three allelic loss of 1p and two allelic loss of 19q, in patients with cerebral tumors which were operated in our Deparment from October 2004 until March 2006. We have detected in blood and tumoral DNA loss of heterozygosity assay for molecular detection using PCR and capillary array electrophoresis of five markers (D1S508, D1S2734, D1S199, D19S412 y D19S219).
Results. Were included in the first part of this study 45 sample of neuroepithelial tissue supratentorial tumors: 29 glioblastoma, 1 gliosarcoma, 7 diffuse astrocytoma grade II, 1 oligoastrocytoma, 3 oligodendroglioma, 1 anaplastic oligodendroglioma, 1 xanthoastrocytoma, 1 dysembryoplastic neuroepithelial tumour and 1 pilocytic astrocytoma. We considered deleted regions identified when allelic ratio (T1/T2)/(N1/N2) was lower than 0.8. Taken together, histological tissue shown deletion in 80% of oligodendroglial tumors, 14% glioblastoma and 14% of diffuse astrocytoma grade II.
Conclusions. Evaluation of 1p/19q allelic status by LOH analysis may provide useful information for guiding clinical and therapeutical decisions with high succes ratio. These results shown why patients with 1p/19q codeletion survive longer, because adjuvant alkylants adds further improvements to standard, surgery and radiotherapy, treatments.
Key words: Cerebral tumors. Gliomas. 1p/19q mutation. Loss of heterozygosity. Alkylating drugs.
Introducción
Entre los tumores de estirpe neuro-epitelial que reconoce la Organización Mundial de la Salud (OMS) en su última revisión del año 200018, los tumores oligodendrogliales forman un subgrupo por sí mismos y también se incluyen sus características en algunos de los gliomas mixtos. Si bien sus hallazgos histopatológicos han sido definidos claramente, en ocasiones es difícil observarlos por su anaplasia o por compartir con astrocitomas el "lecho tumoral" y en estos casos la inmunohistoquímica no consigue unos marcadores definitorios6,9.
Desde la llegada de los marcadores genético-moleculares a los laboratorios de patología hemos podido caracterizar diversos tumores cerebrales y de ellos1,2,23,26 los astrocitomas y oligodendrogliomas han sido objeto de marcado interés. El conocimiento de la oncogénesis nos ha hecho comprender las vías de nacimiento, progresión y malignización de cada estirpe tumoral en particular, pero además, de algunos de estos hallazgos, podremos obtener en un futuro terapias específicas28. El descubrimiento hasta la fecha, prototipo de esta aplicación, ha sido el conocimiento de la mutación 1p/19q en oligodendrogliomas2,3, lo que se relaciona con una mayor sensibilidad a los alquilantes y que se ha intentado extender, con evidentes limitaciones, a otros tumores cerebrales7,14. Incluso podemos aseverar que fruto de esta euforia, tratamos indiscriminadamente con quimioterapia asociada a pacientes con tumores de los que muy posiblemente sólo se beneficien un porcentaje limitado de ellos8,21,25, (10-20% según las series), en términos de supervivencia global. El hecho de que las pérdidas en los cromosoma 1p y 19q estén estrechamente relacionadas, sugiere que ambos actúen sinérgicamente en la oncogénesis de los tumores oligodendrogliales1,21, y en la búsqueda en los cromosomas de los "loci" responsables se han seleccionado algunos de la región telomérica de 1p y 19q1322. Por otro lado, los oligodendrogliomas anaplásicos muestran hasta en un tercio de los casos pérdidas alélicas del cromosoma 9 (9p21), gen CDKN2A, y en el cromosoma 10q, que suelen ocurrir cuando no hay pérdidas en 1p/19q15. La hipermetilación también puede ser un importante mecanismo epigenético por el cual los oligodendrogliomas escapen al control de ciclo celular29. Sin embargo, estos dos últimos hechos no se acompañan de una decisión terapéutica, sino que, hasta la fecha, tienen sólo implicaciones pronósticas.
Es por tanto obligado, para poder tratar lo más selectiva y eficazmente a los pacientes que sufren de un "glioma maligno" y dado lo limitado de nuestros recursos (coste/eficacia), disponer de técnicas que sean muy sensibles y exactas en las determinaciones genético-moleculares y nos permitan dilucidar qué pacientes se beneficiarían de una determinada terapia asociada a la cirugía. De entre ellas, los estudios de marcadores genéticos se pueden realizar mediante tres técnicas principalmente: LOH (pérdida de heterocigocidad por electroforesis capilar), FISH (hibridación in situ fluorescente) y HGC (hibridación genómica comparada), que muestran una elevada concordancia entre sí (73-99%) y son de utilidad para, por ejemplo, determinar la presencia de mutación 1p/9q y lo que ello implica de sensibilidad a la quimioterapia9,16.
Por ello nos planteamos poner a punto la técnica para la detección de aquellos tumores neuro-epiteliales que manifiesten la presencia de mutación 1p-19q por estudio de LOH y que nos permitan seleccionar el tratamiento quimioterápico complementario más adecuado y eficaz.
Material y método
* Muestras tumorales y pacientes. La determinación se realiza en el laboratorio de Inmunología, por comparación a partir del ADN tumoral y de tejido no tumoral, (sangre del paciente) para lo cual hemos de tomar ambas muestras en quirófano y remitirlas de forma inmediata al laboratorio donde van a ser procesadas. La muestra del tumor se selecciona, una vez que tenemos la información de la biopsia intraoperatoria, recogiendo un volumen de unos 2 a 4 cc de la misma zona, evitando zonas macroscópicas de necrosis o vascularizadas. Se tomaron un total de 45 muestras de tumores cerebrales extraídas de pacientes intervenidos quirúrgicamente en nuestro centro (hemos excluido 3 muestras provenientes de otros hospitales), entre octubre de 2004 a marzo de 2006. Los responsables de la técnica de laboratorio desconocen, durante su procesado, el resultado histopatológico de cada muestra. La sangre del paciente se obtiene por venopunción y se recoge directamente en tubos estériles con EDTA como anticoagulante. Las partes de las muestras sobrantes se guardan a -20°C. Todos los pacientes cuyos tumores fueron analizados por este procedimiento, fueron informados de la naturaleza de este estudio y dieron su consentimiento por escrito.* Extracción de DNA. De cada muestra de sangre y tumor se realizó la extracción del DNA. El DNA de la sangre se extrajo mediante el procedimiento del equipo Generation Capture Column Kit (Gentra System)s. El DNA tumoral fue obtenido mediante digestión de piezas en fresco con el equipo QIAamp DNA mini Kit (QIAGEM). En ambos casos se siguieron las instrucciones del laboratorio emisor de los equipos. Se midió la concentración de DNA en el espectrofotómetro y se diluyeron las muestras en agua destilada hasta alcanzar los 30 microg/ml. Todas las muestras se almacenaron a -20°C hasta su análisis.
* Elección de los microsatélites. Los "marcadores" seleccionados fueron todos dinucleótidos -CA- con una puntuación de heterocigocidad en la población general alta, igual o superior a 0.75. Siguiendo a Reifenberger, para el desarrollo de las pruebas clínico-terapéuticas, se debería incluir el estudio de los siguientes microsatélites: para el estudio de la pérdida o delección en el brazo corto del cromosoma 1, deberíamos informar de 3 marcadores: D1S508, D1S199, D1S2734 (todos ellos dentro de la región 1p36). Para el estudio de la pérdida del brazo largo del cromosoma 19, es preciso estudiar los locus: D19S219, D19S412, D19S596 (en la región 19q13.3) (ura1). Nosotros hemos excluido el D19S596 por no cumplir, en la población de nuestro entorno, con la puntuación mínima exigida de tasa de heterocigocidad. Pérdidas de heterocigocidad (LOH) que ocurran fuera de las regiones donde se localizan estos microsatélites (1p36 y 19q13.3) no van a ser detectadas mediante este análisis.

Figura 1. Situación de los microsatélitites
en los cromosomas 1p y 19q.
* PCR y electroforesis capilar. Determinamos la pérdida de heterocigocidad (LOH) en lp y 19q mediante PCR de secuenciación de ADN de microsatélites, que nos permite amplificar las regiones pre-seleccionadas del ADN, definidas por los cebadores o primers. La PCR consta de un número variable de ciclos, generalmente entre 20 y 35, y cada uno de ellos se divide en 3 etapas. Se realizaron en un volumen total de 10 microl., 1 microl. de DNA, 4 microl. de ECOTAQ polimerasa y 5 microl. de primers 2 microM, donde el forward está marcado con un fluorocromo en el extremo 5'. Después de un paso de desnaturalización a 95°C durante 5 minutos en el que se separan las cadenas de DNA, se desarrollan entre 20 y 35 ciclos, cada uno consistente en 30 segundos a 95°C y 1 minuto a 57°C. Seguidamente tienen lugar los ciclos de amplificación en los que por hibridación los primers se unen a la zona de DNA complementaria a la secuencia. Finalmente hay uno de extensión a 72°C durante 10 minutos. En cada ciclo el número de amplificados se duplica, por lo que el incremento es exponencial.
Los cebadores o primers para la amplificación de los microsatélites D1S2734, D1S199, D19S219 y D19S412 ya han sido descritos17. Los del marcador D1S508 se diseñaron en nuestro laboratorio según la secuencia publicada por el Genebank (n° de acceso z24634 y NCBI SNP CLUSTER ID; rs12081216). Todos los primers fueron sintetizados por Sigma (Haverhill, Suffolk, UK) con el extremo 5' del primer sentido de cada pareja marcado con un fluorocromo (TET, HEX o 6 FAM) lo que permite la automatización de la lectura de los marcadores (Tabla 1).
Los productos de la PCR se diluyen en formamida (1vs 12 microl.), junto con 0.5 microl. de marcador de peso molecular TAMRA (que emite fluorescencia en rojo). Se desnaturalizan a 95°C durante 5 minutos y rápidamente se colocan en hielo. Seguidamente se analiza la fluorescencia mediante electroforesis capilar, en un gel de agarosa a 15 V/cm durante un tiempo variable en función del tamaño del fragmento esperado, con el ABIprism 310 genetic analyzer y el software GeneScan, y se guarda el registro gráfico. Este sistema fluorimétrico permite la detección de múltiples loci. Los primers del cromosoma 1 tienen fluorocromos diferentes, lo que permite la lectura simultánea de todos los marcadores de una misma muestra en una sola inyección. Los marcadores del cromosoma 19 de una misma muestra, aunque llevan todos el mismo fluorocromo HEX, también se pueden analizar en una sola inyección ya que los tamaños en pares de bases de los productos de la PCR no se solapan.
Resultados
* Interpretación de los registros gráficos. La intensidad de fluorescencia del alelo largo (mayor número de pares de bases) siempre es menor que la del alelo corto, presumiblemente porque las condiciones de la PCR favorecen la completa replicación del alelo corto durante el primer ciclo de la PCR. Justo delante de cada uno de los alelos vamos a encontrarnos picos de menor intensidad de fluorescencia, a intervalos de -2 nucleótidos, llamados "sombras de bandas", o stutter, que confirman la especificidad de la PCR pues sólo aparecen cuando se amplifican repeticiones de di-, tri- y tetranucleótidos. Son también de utilidad para descartar posibles amplificaciones inespecíficas de la PCR, ya que estas no los presentan. Tradicionalmente la LOH basada en microsatélites, se define como la pérdida relativa de un alelo en el tumor, comparada con tejido sano del mismo paciente. Cuando se presenta LOH el producto de la PCR sólo mostrará una banda de amplificación, correspondiente a uno de los alelos pues el otro se ha deleccionado, mientras que el tejido no tumoral mostrará dos (Figura 2). Es esencial que se recoja tejido sano junto con el tumoral para usarlo como referencia, ya que marcadores homocigotos no serán informativos. Por esta razón también es importante que los marcadores seleccionados tengan ratios de heterocigocidad altos17.
Mientras que, en teoría, la reducción del 100% de la señal alélica es posible, experimentalmente es difícil de conseguir. Esto puede deberse a la naturaleza infiltrativa de esos tumores o a su vascularización, que pueden ocasionar contaminación del DNA por material genético de células no tumorales. Por tanto en el análisis de todas las muestras vamos a considerar que un tumor tiene LOH si presenta una reducción de la señal alélica de al menos un 20% (peak ratio < 0.8)19. Este ratio alélico se calcula como (T1/T2) / (N1/N2) donde T1 y Nl son los picos de fluorescencia correspondientes al alelo de menor tamaño en pares de bases y N2 y T2 los correspondientes al alelo de mayor tamaño (T es tejido tumoral y N es tejido sano), amplificando cada microsatélite en el ADN tumoral y ADN sanguíneo de cada paciente, al tratarse de tejidos en fresco para lo que necesitamos realizar esta comparación, y diremos que existe mutación, LOH en cada microsatélite, bien de los de 1p o 19q, cuando el cociente es < 0.8; no existirá LOH cuando el cociente es > 0.8 (Figura 3).
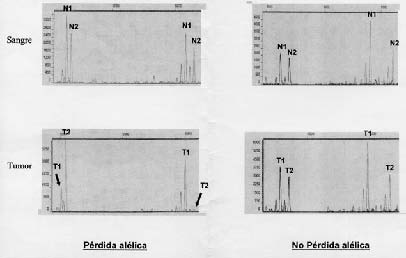
Figura 3. Representación de un electrofe rograma
mostrando un tumor con pérdida alélica (flechas)
junto a otro que no la presenta.
Los picos de fluorescencia de menor peso molecular
corresponden a los alelos del marcador D1S508,
los de mayor peso molecular al D1S2734.
Por delante de cada uno de ellos y a distancia
de 2 nucleótidos encontramos los stutters.
* Pacientes y resultados. Los resultados para cada paciente se muestran en la tabla 2 en la que figuran, junto a su numeración consecutiva, los resultados para los 3 alelos para 1p (D1S508, D1S2734, D1S199), los 2 alelos de 19q (D19S412, D19S219) y el diagnóstico anatomopatológico (AP). El marcador para el D19S596 mostró una tasa de heterocigocidad de tan sólo el 20% en los 10 primeros pacientes estudiados y fue excluido el análisis.
Hemos de considerar que para que exista la mutación, de manera que se exprese el defecto de MGMT (06-metilguanina-DNA-metiltransferasa), responsable último dela sensibilidad a los agentes alquilantes, hemos de demostrar al menos la LOH en 2 alelos del lp y en uno del 19q. Del total de 45 pacientes incluidos en este estudio los resultados histológicos fueron: 29 glioblastomas, 1 gliosar-coma, 7 astrocitomas grado II, 1 oligoastrocitoma, 3 oligodendrogliomas, 1 oligodendroglioma anaplásico, 1 xantoastrocitoma, 1 tumor neuroepitelial disembrioplásico y 1 as-trocitoma pilocítico. De ellos resultaron presentar la mutación (LOH) según los criterios antes expuestos: un 80% de tumores oligodendrogliales (un oligoastrocitoma, un oli-godendroglioma anaplásico y 3 de los cuatro oligodendrogliomas grado II), 14% de glioblastomas (4 de 29 casos) y 14% de astrocitomas fibrilares grado II (1 de 7 pacientes).
Discusión
La técnica de detección de LOH en alelos de los cromosomas lp y 19q es posible llevarla a cabo en nuestros laboratorios sin un coste excesivo, pero precisa de personal cualificado y experimentado en el manejo de técnicas en biología molecular, que dominen las técnicas de amplificación de ADN, electroforesis, lectura de fluorescencia, etc... Los resultados recientemente publicados por parte de la RTOG (9402) y la EORTC en ensayos randomizados fase III, sobre pacientes con oligodendrogliomas anaplásicos y oligoastrocitomas anaplásicos, muestran que la codelección de los cromosomas 1p y 19q tienen un impacto significativo sobre la supervivencia de los pacientes, hasta tal punto que los próximos ensayos clínicos caracterizarán a los gliomas anaplásicos basados en el genotipo más que en las características histopatológicas5,27.
Los resultados que hemos obtenido son semejantes a los referidos en la literatura por parte de centros que las han aplicado con fines bien experimentales o clínicos. Manifiestan la LOH casi el 100% de los oligodendrogliomas, hasta un 15-20% de Astrocitomas grado II y sólo un 10% de los Glioblastomas4,14,15,22. Esto justificaría la alta tasa de sensibilidad a los alquilantes (quimioterapias con PCV o temozolamida) de los oligodendrogliomas, la respuesta ocasional de algunos astrocitomas de bajo grado, posiblemente pues realmente se trate de astrocitomas que filogenéticamente son de estirpe oligodendroglial más que astrocitaria, y la tasa de respuestas con aumentos de supervivencia global cercana al 10% que presentan los pacientes con glioblastomas8,25. Otra posible explicación a estos hallazgos es que en dos de estos tumores, el astrocitoma de bajo grado y un glioblastoma, se ha encontrado que la delección era parcial en 1p, hallazgo este que ocurre con más frecuencia en astrocitomas que en oligodendrogliomas, lo que se atribuye a la mayor inestabilidad genética de estas neoplasias11.
Respecto a la técnica elegida para mostrar la existencia de mutación en 1p/19q, nos decidimos a emplear la LOH por diversos motivos: el primero es que los Dres M.J. Bello y J.A. Rey, expertos en técnicas genético-moleculares sobre tumores cerebrales, nos la recomendaron como la más específica2; en segundo lugar, teníamos la oportunidad de contar en nuestro hospital con expertos en estas técnicas dentro del Servicio de Inmunología; por último. podíamos emplearla en tejido en fresco y luego contrastarla con la misma técnica en ADN extraído del tumor en bloques de parafina, fase de estudio en la que nos encontramos en la actualidad, junto con la determinación simultánea por FISH por parte del Servicio de Anatomía Patológica de nuestro centro. Aunque otros autores ya han señalado la alta tasa de concordancia de las técnicas genético-moleculares9,24, nosotros queremos evidenciar cuál es esta en nuestros laboratorios, incluso abriendo la posibilidad futura de emplear más de una para su confirmación, o dependiendo del tipo tumoral que estemos analizando.
A partir de los resultados obtenidos podemos planteamos dos aplicaciones clínicas inmediatas: la primera es que la demostración de la LOH de 1p/19q nos va a indicar qué pacientes, que sin esta determinación no trataríamos con alquilantes, se pueden beneficiar de este tratamiento, por ejemplo un astrocitoma fibrilar irresecable. En segundo lugar nos permite analizar retrospectivamente, si se corresponde la supervivencia alcanzada con tratamiento quimioterápico complementario en aquellos pacientes con glioblastomas y oligodendrogliomas que presenten la LOH o no, pero que tratamos sistemáticamente en nuestro centro con alquilantes8, de manera que apreciemos si el aumento de supervivencia global es sólo a costa de los que tienen la LOH, sin que el resto se beneficie, lo que nos obligaría a replanteamos el tratamiento que aplicamos, como han sugerido otros autores10,12.
En un futuro irán surgiendo tratamientos complementarios a la cirugía más específicos, como anticuerpos monoclonales, inhibidores de kinasas, etc..., que nos obligarán a tener dispuestas técnicas de laboratorio para determinar a estos subgrupos de pacientes con tumores cerebrales13. Recientemente, la escasa aplicación clínica que tenía en el subgrupo de glioblastomas con mutaciones del cromosoma 10 la determinación de mutaciones en EGFR, parece haber encontrado en unos marcadores moleculares, variante III (EGFRvIIl) y la pérdida simultánea del GOS PTEN, a aquellos que identifican a los pacientes que serían sensibles al tratamiento con los inhibidores de tirosin-kinasa (erlotinib o gefitinib)20.
Conclusiones
La determinación de la LOH en lp/19q es un método posible para desarrollarlo en los laboratorios de nuestros hospitales que dispongan de técnicas en biología molecular, con una alta especificidad y una aplicación directa e inmediata que redundará en beneficio del paciente. De ella se benefician no sólo los pacientes con tumores oligodendrogliales, sino un porcentaje variable, entre el 10-20 %, de otros tumores de estirpe neuroepitelial que muestran la LOH.
Agradecimientos
A los Dres. López-Escobar, López López, García Vázquez, Gómez Cárdenas y Rodríguez Peña por colaborar en la toma de muestras durante los procedimientos quirúrgicos.
Bibliografía
1. Bello, M.J.; Leone, P.E.; Vaquero, J.; de Campos, J.M.; Kusak, M.E.et al.: "Allelic loss at 1p and 19q frequently occurs in association and may represent early oncogenic events in oligodendroglial tumors". Int.J.Cancer 1995; 64: 207-210. [ Links ]
2. Bello, M.J.; Rey, J.A.: "Oligodendrogliomas: biología molecular". Neurocirugía 1999; 10: 381-386.
3. Cairncross, J.G.; MacDonald, D.; Ludwin, S. et al: "Chemotherapy for anaplastic oligodendroglioma". J.Clin.Onco1.1994; 12: 2013-2021.
4. Cairncross, J.G.; Ueki, K.; Zlatescu, M. et al: "Specific genetics predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. Current treatment of oligodendrogliomas". J. Natl.Cancer Institut.1998;90: 1473-1479.
5. Cairncross, J.G.; Berkey, B.; Shaw, E. et al: "Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402". J. Clin. Oncol. 2006; 24: 2707-2714.
6. Coca, S.; Vaquero, J.: "Oligodendrogliomas: Concepto anatomopatológico y características biológicas generales". Neurocirugía, 1999; 10: 373-377.
7. Engelhard, H.H.; Stelea, A.; Cochran, E.J.: "Oligodendroglioma: pathology and molecular biology". Surg. Neurol. 2002; 58: 111-117.
8. Gil-Salú, J.L.; Román, P. ; Benítez, E. et al : "Análisis de la supervivencia global al añadir temozolamida al tratamiento quirúrgico y radioterápico en pacientes con Glioblastoma multiforme". Neurocirugía 2004; 15: 144-150.
9. Gupta, M.; Djalilvand, A.; Brat, D.: "Clarifying the diffuse gliomas: a update on the morphologic features and markers that discriminate oligodendroglioma from astrocytoma". Am. J. Clin. Pathol. 2005; 124: 755-768.
10. Hamlat, A.; Saikali, S.; Chaperon, J.; Le Calve, M.; Gedouin, D. et al: "Oligodendroglioma: clinical study and survival analysis correlated with chromosomal anomalies". Neurosurg. Focus 2005; 19 (5): E15 (1-9).
11. Hartmann, C.; Mueller, W.; Lass, U.; Kamel-Reid, S.; von Deimling, A.: "Molecular genetic analysis of oligodendroglial tumors". J.Neuropathol. Exp. Neurol. 2005; 64: 10-14.
12. Hegi, M.E.; Diserens, A.C.; Gorlia, T. et al: "MGMT gene silencing and benefit from temozolomide in glioblastoma". N. Engl. J. Med. 2005; 352, n°10: 997-1003.
13. Hill, Ch.; Hunter, S.B.; Brat, D.J.: "Genetic markers in Glioblastoma: prognostic significance and future therapeutic implications"; Adv. Anat. Pathol. 2003; 10: 212-217.
14. Ino, Y.; Zlatescu, M.C.; Sasaki, H. Et al.: "Long survival and therapeutic responses in patients with histologically disparate high-grade gliomas demostrating chromosome 1p loss". J.Neurosurg. 2000; 92: 983-990.
15. Ino, Y; Betensky, R.A.; Zlatescu, M.C. et al: "Molecular subtypes of anaplastic oligodendrogliomas: implications for patient management at diagnosis". Clin. Cancer Res. 2001; 7: 839-845.
16. Kelley, T.W.; Tubbs, R.R.; Prayson, R.A.: "Molecular diagnostic techniques for the clinical evaluation of gliomas". Diagn. Mol. Pathol. 2005; 14: 1-8.
17. Kim, S.H.; Kim, H.; Kim, T.S.: "Adequate microsatellite markers for 1p/lq loss of heterozygosity of oligodendroglial tumors in Korean patients". Korean J. Pathol. 2005; 39: 22-33.
18. Kleihues, P; Cavenee, W.K.: "World Health Orga nization Classification of tumours; Pathology and Genetics; Tumours of the nervous system". IARC Press; Lyon 2000.
19. Medintz, LL.; Lee, C.C.; Wong, W.W.; Pirkola, K. Sidransky, D.; Mathies, R.A.: "Loss of heterozygosity assay for molecular detection of cancer using energy-transfer primers and capillary array electrophoresis". Genome Res. 2000; 10: 1211-1218.
20. Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I. et al: "Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors". N. Engl. J. Med. 2005; 353: 20122024.
21. Nutt, C.L.: "Molecular genetics of oligodendrogliomas: a model for improved clinical management in the field of neurooncology". Neurosurg. Focus. 2005; 19: 1-9.
22. Reifenberger, G. And Louis, D.N.: "Oligoden droglioma: toward molecular definitions in diagnostic neuro oncology". J. Neuropathol. and Exp. Neurol. 2003; 62: 111-126.
23. Rey, J.A.; Bello, M.J.; De Campos, J.M. et al: "Chromosomal patterns in human malignant astrocytomas". Cancer Genet. Cytogenet., 1987; 29: 201-221.
24. Smith, J.S.; Alderete, B.; Minn, Y.; Borell, T.J.; Perry, A. et al: "Localization of common deletion regions on 1p and 19q in human gliomas and their association with histological subtype". Oncogene 1999; 18: 4144-4152.
25. Stupp, R.; Mason, W.P.; van de Bent, M. Et al: "Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma". N. Engl. J. Med. 2005; 352, n°10: 987-996.
26. Van den Bent, M.J.; Chinot, O.L.; Cairncross, G.: "Recent developments in the molecular characterization and treatment of oligodendroglial tumors". Neuro-oncology, 2003;5: 128-138.
27. Van den Bent, M.J.; Carpentier, A.F.; Brandes, A.A. et al: "Adjuvant procarbazine, lomustine, and vincristine improves progressión-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial". J.Clin. Oncol. 2006; 24: 2715-2722.
28. Vaquero, J. y Martínez, R.: "Nuevas perspectivas en el tratamiento biológico de los gliomas". Neurocirugia, 1990; 1: 197-201.
29. Watanabe, T.; Nakamura, M.; Yonekawa, Y. et al: "Promoter hypermethylation and homozygous deletion of the p14ARF and pl6INK4a genes in oligodendrogliomas". Acta Neuropathol. 2001; 101: 185-189.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. J.L. Gil-Salú.
Apdo. Correos 167. 11100.
San Fernando. Cádiz.
Recibido: 10-10-06.
Aceptado: 20-11-06
Abreviaturas. ADN: ácido desoxi-ribonucleíco. PCV: procarbazina, lomustina (CCNU) y vincristina. EORTC: European Organisation for Research and Treatment of Cancer. GOS: gen onco-supresor. PCR: reacción de cadenas de polimerasas. RTOG: Radiation Therapy Oncology Group. U.K.: Reino Unido
Comentario al artículo Análisis de la presencia de mutaciones por pérdida de heterocigocidad de lp/l9q en tumores cerebrales de estirpe glial, de J.L. Gil Salú y cols.
En el trabajo presentado por Gil-Salú y cols., se estudia la presencia de mutaciones cromosómicas 1p y 19q en muestras de tumores cerebrales, analizando la pérdida de heterocigocidad. Sus resultados señalan que, al menos en su serie, existen mutaciones en el 80% de los tumores oligodendrogliales, en el 14% de los glioblastomas y en el 14% de los astrocitomas de bajo grado. Estimamos que los autores deben ser felicitados, no sólo por la claridad del artículo, sino por su indudable interés y oportunidad, en un momento en que se está generalizando en neuro-oncología el empleo de nuevos agentes quimioterápicos y se hace preciso seleccionar los casos en los cuales el empleo de sustancias "alquilantes" puede tener mayor efectividad. La lectura de artículos como éste muestra, una vez más, que el tratamiento de los tumores cerebrales debe ser hecho desde una perspectiva multidisciplinar y que hoy día, el neurocirujano que se dedique a este campo no debe limitarse a conocer perfectamente la técnica quirúrgica, sino que también debe interesarse por los aspectos de la biología molecular de los diferentes tipos de tumores cerebrales y, en colaboración con otros especialistas, debe tratar de identificar nuevos factores pronósticos que permitan un mejor tratamiento de nuestros pacientes.
J. Vaquero
Madrid

