










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkFarmacia Hospitalaria
versión On-line ISSN 2171-8695versión impresa ISSN 1130-6343
Farm Hosp. vol.38 no.3 Toledo may./jun. 2014
https://dx.doi.org/10.7399/FH.2014.38.3.7314
Nuevos fármacos en el abordaje terapéutico de la hepatitis C
New drugs in the treatment of chronic hepatitis C
R. Jiménez Galán, Á. Albacete Ramírez, P. Monje Agudo, Y. Borrego Izquierdo y R. Morillo Verdugo
Hospital Universitario de Valme. España.
Dirección para correspondencia
RESUMEN
Objetivos: Analizar la eficacia y seguridad de los nuevos agentes antivirales directos (AAD) que formaran parte del arsenal terapéutico para el tratamiento de la hepatitis C.
Método: Se realizó una búsqueda en la base de datos electrónica PubMed, de artículos publicados Febrero de 2014 que cumplieran los siguientes criterios: ensayos clínicos (EC) en fase II o III y cuyos objetivos fueran evaluar la eficacia y seguridad de nuevas generaciones de inhibidores de la proteasa (IP) frente al VHC, excluyendo con boceprevir y telaprevir.
Resultados: Se incluyeron de 24 EC que incluyen asociaciones de AAD con ribavirina (RBV) y con o sin peginterferon (PegINF) y asociaciones de varios AAD. Los resultados de daclatasvir con PegINF y RBV no han sido muy satisfactorios. Por el contrario, sofosbuvir es activo en todos los genotipos virales y permite ser administrado en regímenes libres de PegINF. Alrededor del 90% de los pacientes naïve alcanzan respuesta viral sostenida (RVS), y no llegan al 80% en pretratados. En cuanto a la segunda generación de IP NS3/4A, destacar a simeprevir, con respuestas próximas al 90% en pacientes naïve y cercanas al 80% en pretratados. Entre las combinaciones de AAD evaluadas, sofosbuvir y daclatasvir y sofosbuvir y ledipasvir alcanzan el 100% de respuesta en no respondedores a triple terapia con boceprevir y telaprevir.
Conclusiones: Las nuevas generaciones de AAD frente al VHC van a suponer un aumento de las tasas de curación en todos los subtipos de pacientes, a través de regímenes más sencillos y mejor tolerados.
Palabras clave: Hepatitis C crónica; Terapia; Agente antiviral directo.
ABSTRACT
Objectives: To analyze the efficacy and safety of the new direct antiviral agents (DAA) that will become the new therapeutic arsenal for the treatment of hepatitis C.
Methods: We carried out a research in the electronic database with the following criteria: phase II and III clinical trials (CT) published until February 2014. The Mesh term used was "chronic hepatitis C" and "therapy". Studies with boceprevir or telaprevir were excluded. For the analysis of efficacy, we evaluated the rate of Sustained Viral Response (SVR), and for the safety, side effects and safety-related discontinuations were analyzed.
Results: We included 24 CT that include associations with ribavirine (RBV) with or without peginterferon (PegINF) and associations of several DAA. The results associated of daclatasvir with PegINF and RBV have not been very successful. On the contrary, sofosbuvir presents activity in all viral genotypes . Sofosbuvir may be administered in free PegINF regimens. Around 90% of naïve patients achieve sustained virological response (RVS) and 80% in previously treated. In relation to second wave of NS3/4A protease inhibitors, simeprevir has achieved RVS in 90% of naïve patients and close to 80% in previously treated. The main combination of DAA were sofosbuvir and daclatasvir and sofosbuvir and ledipasvir. Both have achieved SVR in 100% of patients who previously had virological failure after receiving a protease inhibitor regimen with boceprevir or telaprevir.
Conclusions: The new generation of AAD for the treatment of hepatitis C will lead to higher response rates in all subtypes of patients with lower complexity regimens and better tolerated.
Key words: Chronic hepatitis C; Therapy; Direct acting antiviral.
Introducción
El virus de la hepatitis C (VHC) constituye un problema de salud a nivel mundial afectando a más de 170 millones de personas en el mundo. Es la principal causa de cirrosis hepática y hepatocarcinoma. En conjunto, estos datos sugieren que el VHC es responsable de aproximadamente un millón de muertes al año1.
La evolución del tratamiento en los últimos años ha sido francamente espectacular. Hasta el año 2011 la terapia estándar y única disponible consistía en la biterapia de interferon pegilado (PegINF) alfa-2a o alfa 2b junto con ribavirina (RBV) administrada durante 24 o 48 semanas2. Sin embargo, con esta terapia, sólo el 50% de los pacientes infectados por el genotipo 1 alcanzaban Respuesta Viral Sostenida (RVS) y con una tasa de efectos adversos importantes. En el año 2011, cambió por completo el escenario terapéutico del tratamiento de la hepatitis C con la aparición de la primera generación de inhibidores de la proteasa del VHC: Boceprevir (BOC) y Telaprevir (TVR)3. Ambos fármacos han sido aprobados por la la EMA (European Medicines Agency)4 y por la FDA (U.S Fud and Drug Administration)5 para el tratamiento de pacientes naíve y previamente tratados infectados por el genotipo 1. Esta terapia es lo que conocemos como la "triple terapia" ya que requiere su administración en combinación con PegINF y RBV. La aparición de los Agentes Antivirales directos (AAD) permite alcanzar tasas de RVS significativamente mayores6,7,8 comparado con la biterapia2,9 (59-73% vs 23-50%) ofreciendo así la oportunidad de curación a pacientes que tienen muy pocas probabilidades de responder al tratamiento con PegIFN y RBV o que previamente habían fracasado a la biterapia. Sin embargo, a pesar de los grandes beneficios clínicos conseguidos con la aparición de los inhibidores de la proteasa, también conlleva una serie de inconvenientes; por una lado la aumento considerable de efectos secundarios comparado con la biterapia con PegIFN+ RBV10, que además éstos pueden resultar de carácter grave y afectar de forma importante a la calidad de vida del paciente , y por otro lado la necesidad de administrar estos fármacos junto con PegIFN y RBV, suponiendo esto un incremento importante de la complejidad del régimen terapéutico.
Actualmente están apareciendo nuevas generaciones de inhibidores de la proteasa del VHC. Algunos de ellos, como es el caso de sofosbuvir y simeprevir han sido aprobados recientemente por la FDA , otros se encuentran ya en estudios en fase III. Este nuevo panorama terapéutico pretende por una lado, mejorar las tasas de RVS en aquellos pacientes con una perfil desfavorable, como son los no respondedores a tratamiento previo y cirróticos, y por otro lado, el desarrollo de regímenes menos complejos y con una tasa inferior de efectos adversos. Paralelamente, estos nuevos fármacos no sólo están siendo evaluados en pacientes con genotipo 1, sino también en otros genotipos con el fin de buscar regímenes mejor tolerados libres de interferon (INF).
Por tanto, el objetivo de este estudio, es analizar la eficacia y seguridad de los nuevos AAD que un futuro muy próximo formaran parte del arsenal terapéutico para el tratamiento de la hepatitis C.
Material y métodos
Se realizó una búsqueda en la base de datos electrónica PubMed de artículos publicados sin restricción de idioma hasta Febrero de 2014 en los que se evaluará la eficacia y seguridad de nuevas generaciones de AAD dirigidos frente al VHC. Los criterios de inclusión fueron: diseño del estudio (ensayos clínicos (EC) en fase II o III) y cuyos objetivos fueran evaluar la eficacia y seguridad de nuevas generaciones de inhibidores de la proteasa (IP) frente al VHC. Los criterios de exclusión fueron: EC fase I, estudios con fármacos inhibidores de la proteasa de primera generación (boceprevir y telaprevir).
Para llevar a cabo la búsqueda se utilizó como término MESH "chronic hepatitis C", "therapy" y posteriormente se limitó la búsqueda por EC fase II y III y por fecha.
Para el análisis de la eficacia, la variable principal considerada fue la tasa de RVS: porcentaje de pacientes con carga viral negativa 12 o 24 semanas después de finalizar el tratamiento antiviral. La eficacia fue analizada en los distintos subgrupos de pacientes: genotipos virales, pacientes naïve o previamente tratados. También se analizaron factores predictores de RVS: presencia o no de cirrosis , genotipo CC vs no CC de la IL28B, genotipo viral 1a vs 1b. En cuanto a la seguridad, se analizaron los efectos adversos más frecuentes asociados al tratamiento con el AAD, así como la tasa de discontinuación por este motivo. Finalmente, también se determinó la tasa de repuntes virológicos y recidiva y su asociación con el desarrollo de resistencias a cada uno de los nuevos fármacos.
Resultados
Un total de 133 artículos fueron filtrados tras la búsqueda. De los cuales, sólo 24 cumplieron los criterios de selección establecidos.
Las características, diseño, población, así como las tasas globales de RVS de los mismos se resumen en la tabla 1. En cuanto al mecanismo de acción que presenta cada AAD, en la figura 1 se muestra el punto exacto en el que actúan inhibiendo la replicación viral.
Inhibidores de la polimerasa NSSA
Daclatasvir
Daclatasvir (BMS-790052) es el primer AAD en su clase que actúa bloqueando la NS5A, proteína clave en la replicación viral del VHC11.
- Eficacia de daclatasvir en pacientes naïve. La eficacia de daclatasvir en combinación con pegINF y RBV ha sido evaluada sólo en pacientes naïve con genotipo 1 en un EC en fase Il12. Este estudio incluyó a 48 que fueron asignados aleatoriamente a recibir daclatasvir 3 mg, 10 mg o 60 mg en una dosis diaria (QD) o placebo en combinación con PegINF y RBV durante 48 semanas. Las características fueron similares en los distintos grupos, salvo en los pacientes que recibieron daclatasvir 60 mg, la proporción de pacientes con genotipo no CC de la IL28B fue mayor. El objetivo principal de este estudio fue determinar la tasa de respuesta virológica rápida extendida (RVRe) , es decir determinar la proporción de pacientes que alcanzaban carga viral indetectable desde la semana 4 hasta la semana 12 de tratamiento. Como variables secundarias también se determinó la tasa de RVS. Este estudio fue descriptivo, por tanto no pretendía demostrar diferencias estadísticamente significativas entre las distintas terapias evaluadas.
La proporción de pacientes que alcanzaron RVRe fue del 42%, 83% y 75% para las dosis de daclatasvir de 3 mg, 10 mg y 60 mg, respectivamente frente al 8% en el grupo placebo. Mientras que la tasa de RVS en la semana 24 post tratamiento (RVS24) fueron del 42%, 83% Y 83% para las dosis de daclatasvir de 3, 10 y 60mg , respectivamente comparada con el 25% en el grupo placebo.
- Seguridad del tratamiento con daclatasvir. El 16% de los pacientes discontinuaron el tratamiento por motivos de seguridad. Los efectos secundarios más frecuentes asociados a daclatasvir fueron dolor de cabeza, fatiga y ageusia.
- Variaciones genotípicas asociadas con resistencia. En algunos de los pacientes que presentaron fracaso virológico fueron detectados polimorfismos asociados con resistencia a daclatasvir.
Inhibidores de la polimerasa NSSB
Sofosbuvir
Sofosbuvir (GS-7977) es un análogo de nucleótido que actúa inhibiendo de forma selectiva la polimerasa NS5B (Figura 1), enzima clave en el ciclo de replicación del VHC. Sofosbuvir es un profármaco del 2-desoxi-2-fluoro-2-C-metiluridina monofosfato, que se transforma en el interior de los hepatocitos en su forma activa, provocando así la terminación de la replicación del genoma viral13. Los resultados de eficacia y seguridad han llevado a su reciente aprobación por la FDA14 en pacientes naive y pretratados con genotipos 1, 2, 3 y 4 del VHC. Como hecho histórico en el tratamiento de la hepatitis C, por primera vez se aprueba una terapia frente al VHC libre de PegINF, aunque sólo en pacientes con genotipos 2 y 3, ya que aquellos infectados por el genotipo 1 o 4 si requieren su administración.
- Eficacia de Sofosbuvir en pacientes naïve. La eficacia y seguridad de sofosbuvir en pacientes naïve ha sido evaluada en todos los genotipos virales. El primer EC en fase II publicado fue el estudio ELECTRON15 (Tabla 1), en el que se evaluaron distintos regímenes con sofosbuvir, incluyendo aquellos libres de PegINF y la monoterapia con sofosbuvir (Tabla 2). En este estudio se incluyeron pacientes no cirróticos con genotipo 1,2 y B. Las tasas de RVS en la semana 12 post tratamiento fueron muy elevadas en todos los subgrupos de pacientes (Tabla 2), 84-100%, a excepción del grupo que recibió monoterapia con sofosbuvir (el 60% alcanzó RVS) y la cohorte de pacientes genotipo 1 previamente tratados (10% presentaron RVS).
Similares tasas de respuestas (85-92%) fueron alcanzadas en otro EC en fase II16 también realizado en pacientes no cirróticos con genotipos 1, 2 y 3 (Tabla 3). A diferencia del estudio ELECTRON15, en éste todos los pacientes recibieron triple terapia con sofosbuvir, PegINF y RBV durante 12 semanas seguido de otras 12 semanas de PegINF y RBV en los pacientes con genotipo 2 y 3, mientras que en los pacientes con genotipo 1 se llevó a cabo la terapia guiada por respuesta (TGR), de manera que aquellos pacientes que alcanzaron RVRe recibían 12 semanas adicionales con pegINF y RBV y el resto 36 semanas. La cohorte de pacientes con genotipo 1 comparó distintas dosis de sofosbuvir (200 vs 400 mg al día) y fue controlada con placebo, mientras que la otra no. La respuesta fue similar independientemente de la dosis de sofosbuvir y de la realización de TGR (90-92%). Por otro lado, en los pacientes con genotipo 1 se observó que la RVS en la semana 24 post tratamiento se redujo con respecto a la semana 12, mientras que en el grupo placebo se mantuvo igual (Tabla 3).
Los resultados del estudio ATOMIC17, fueron consistentes con los obtenidos en estudios previos. Este otro EC en fase II fue llevado a cabo en pacientes no cirróticos con genotipos 1,4, 5 y 6. Las tasas de RVS en la semana 12 y 24 post tratamiento fueron similares en los distintos grupos de tratamiento (90-93% y 87-89%), independientemente de la duración del tratamiento y la realización de TGR (Tabla 4).
Con respecto a los resultados de sofosbuvir en fase III en pacientes naïve, disponemos de los resultados de dos estudios: el estudio FISSION y NEUTRINO18.
El estudio FISSION18, fue llevado a cabo en pacientes con genotipos 2 y 3 e incluyó una pequeña proporción de pacientes cirróticos (Tabla 1). En este estudio se comparó la eficacia de la biterapia con sofosbuvir 400 mg al día (QD) y RBV a dosis estándar (1000 mg o 1200 mg en función del peso) durante 12 semanas frente a pegINF alfa 2a (180 mcg/semana) y RBV (dosis diaria de 800 mg) durante 24 semanas. La variable principal de eficacia fue la RVS12.
El tratamiento con sofosbuvir y RBV demostró ser no inferior al régimen con PegINF y RBV (p < 0,001), alcanzándose una tasa de RVS12 del 67% en ambos grupos. En los pacientes que recibieron sofosbuvir, la infección por genotipo 2 y la ausencia de cirrosis se correlacionaron de forma significativa con la RVS. Como aspecto a destacar de este estudio es que el grupo control recibió una dosis de RBV inferior.
Por otro lado, el estudio NEUTRINO18, incluyó a 327 pacientes naïve con genotipos 1, 4, 5 y 6 a recibir triple terapia con sofosbuvir (400 mg al día), RBV y PegINF alfa 2a durante 12 semanas. El endpoint de este estudio fue la RVS12, para demostrar la superioridad del régimen con triple terapia era superior a las tasas de RVS históricamente obtenidas con pegINF y RBV (60%).
El 89% de los pacientes fueron genotipo 1, el 9% genotipo 4 y el restante genotipo 5 o 6. El 17% de los pacientes fueron cirróticos y el 71% eran portadores del genotipo no CC para la IL28B. Los resultados virológicos demostraron la superioridad del régimen con sofosbuvir, ya que el 90% alcanzaron RVS12 (p < 0,001). La respuesta en función del genotipo fue la siguiente: 89% genotipo 1, 96% en genotipo 4 y 100% en genotipos 5 y 6. Al igual que el estudio FISSION, la ausencia de cirrosis fue asociada de forma significativa con la RVS (92% vs 80%), junto con el genotipo de la IL28B (98% en pacientes con genotipo CC vs 87% genotipo no CC).
Las características basales de los pacientes incluidos en los estudios previamente presentados son mixtas. Sólo los EC en fase III (FISSION y NEUTRINO18) incluyeron una pequeña proporción de pacientes cirróticos (17-20%). Existen datos publicados de un EC en fase II19 que incluye a 50 pacientes naïve genotipo 1 con características desfavorables (70% genotipo 1a, 83% raza negra, 48% IMC > 30 kg/m2, 81% genotipo no CC de a IL28B, 23% F3-F4, 62% Carga viral basal > 800.000 UI/mL). Los pacientes fueron aleatorizados a recibir sofosbuvir (400 mg QD) y RBV a dosis estándar o sofosbuvir a igual dosis y RVB a dosis bajas (600 mg al día) durante 24 semanas. En primer lugar las tasas de RVS24 fueron inferiores a las obtenidos en otros estudios18,20 (68% vs 85-89%). Por otro lado, en este estudio se pone de manifiesto el papel clave que juega la RBV en la terapia, debido al incremento de la respuesta observado en los pacientes que recibieron RBV a dosis estándar (68% vs 48%), aunque está diferencia no alcanzó la significación estadística.
- Eficacia de Sofosbuvir en pacientes previamente tratados. Por un lado, el estudio POSITRON20 es un EC doble ciego y controlado con placebo que incluyó a pacientes con genotipos 2 y 3 sin opciones de tratamiento y previamente tratados que habían interrumpido el tratamiento con interferon (INF) por distintos motivos (efectos adversos, decisión del paciente, o intolerancia/incompatibilidad) fueron aleatorizados en proporción 3:1 a recibir tratamiento con Sofosbuvir (400 mg QD) y RBV o placebo. Un 16% de los pacientes incluidos fueron cirróticos.
La tasa de RVS12y RVS24 fue del 78% (IC 95%; 7283) en el grupo tratado con sofosbuvir, comparado con el 0% en el grupo placebo (p < 0,001). Al igual, que en el estudio FISSION18, el genotipo 2 fue identificado como un factor predictor de RVS, ya que el 93% de los pacientes con genotipo 2 obtuvieron RVS frente al 61% de los pacientes con genotipo 3. Además en este estudio, la cirrosis fue asociada con menores tasas de respuesta, especialmente en los pacientes con genotipo 3 (tasas de RVS del 68% en no cirróticos vs 21% en cirróticos), mientras que los pacientes con genotipo 2 no se vieron afectados por este efecto (tasas de RVS del 92% en no cirróticos y 94% en cirróticos).
Por otro lado, en el estudio FUSION20 se incluyeron a pacientes con genotipos 2 y 3 no respondedores al tratamiento previo con PegINF y RBV En este estudio, la presencia de cirrosis tampoco fue considerada como un criterio de exclusión. Los pacientes fueron aleatorizados en proporción 1:1 a recibir 12 semanas de sofosbuvir y RBV, seguido por 4 semanas de placebo o 16 semanas de tratamiento con sofosbuvir y RBV. La tasa de RVS fue significativamente superior (p < 0,001) en el grupo tratado con sofosbuvir y RBV durante 16 semanas (73% vs 50%).
En este estudio, al igual que en los estudios POSITRON20 y FISSION18, el genotipo 3 fue asociado con tasas de respuesta significativamente menores, ya que el 86% y 94% presentaron RVS en los pacientes con genotipo 2 que recibieron tratamiento 12 y 16 semanas, respectivamente, frente al 30% y 62% en los pacientes con genotipo 3.
La presencia de cirrosis también estuvo asociada con menores tasas de respuesta, aunque esto ocurrió especialmente en pacientes con genotipo 3 que recibieron 12 semanas de tratamiento.
- Seguridad del tratamiento con Sofosbuvir. La tasa de discontinuación por efectos adversos (EA) en los grupos tratados con sofosbuvir fue baja (1-4%) en todos los estudios. La tasa de EA de grado 3 o superior también fue baja (1-15%) y entre 1-5% fueron de carácter relevante. Los más frecuentes detectados en los distintos EC (Tabla 5) fueron fatiga, dolor de cabeza, náuseas, insomnio, y prurito. En el estudio POSITRON20 se observaron elevaciones de las transaminasas en el 22%, aunque este evento no fue observado en el resto de estudios.
- Variaciones genotípicas asociadas con resistencia. En ninguno de los estudios hubo casos de repuntes virológicos. La tasa de recidiva tras finalizar el tratamiento fue variable, oscilando desde un 3-4% en el estudio ATOMIC17 hasta un 20-37% en los estudios FUSION y POSITRON20. Sólo en el estudio ELECTRON15 fueron detectadas mutaciones en el gen NS5B de la polimerasa que podían conferir resistencia al tratamiento con sofosbuvir. Sin embargo, esta mutación fue detectada en todos los pacientes que presentaron recaída tras finalizar el tratamiento y que habían recibido monoterapia con sofosbuvir.
Inhibidores de la NS3/4A
Faldaprevir
Faldaprevir (BI 201335) es un potente inhibidor de la protesa HCV NS3/4A (Figura 1) con un perfil farmacocinético que permite una única administración diaria21. Faldaprevir ha sido evaluado en pacientes con genotipo 1 en varios EC en fase II (SILENC-122 y 223 y otro realizado en una cohorte de pacientes japonesa 24) y también en EC en fase III25 (STARTverso1, 2, 3 y 4) de los que sólo se disponen resultados preliminares, ya que todavía éstos no han sido publicados.
- Eficacia de Faldaprevir en pacientes naive. En el estudio SILENC-122 se evaluaron distintos regímenes de triple terapia con faldaprevir frente a placebo cuya duración en todos los casos fue de 48 semanas: grupo 1)placebo, PegINF y RBV durante 24 semanas seguido de 24 semanas de PegINF y RBV; grupo 2) período de lead-in de 3 días con PegINF y RBV seguido de 24 semanas de triple terapia (Faldaprevir 120 mg QD, PegINF y RBV) y posteriormente 24 semanas de biterapia con PegINF y RBV; grupo 3) el mismo régimen que el anterior, pero con una dosis de faldaprevir de 240 mg QD; y grupo 4) 24 semanas de triple terapia (faldaprevir 240 mg QD, PegINF y RBV) seguido de 24 semanas de biterapia. En los pacientes tratados con faldaprevir 240 mg (con o sin lead-in) se realizó TGR, de modo que aquellos que alcanzaron RVR mantenida (ARN-VHC indetectable en la semana 4 y posteriormente desde la 8-20) fueron aleatorizados a cesar el tratamiento en la semana 24 o bien a continuar hasta la semana 48 con biterapia.
La tasa de RVS en los grupos tratados con faldaprevir 120 mg (con lead-in), 240 mg con y sin lead-in fueron del 72%,72% y 84%, respectivamente, frente al 56% alcanzado en el grupo de placebo. Hubo diferencias estadísticamente significativas entre los grupos que recibieron 240 mg de faldaprevir (con o sin lead-in) comparado con placebo, mientras que el grupo tratado con 120 mg de faldaprevir con lead-in no se alcanzó la significación estadística.
En cuanto a la TGR en los pacientes tratados con faldaprevir, aunque no hubo diferencias significativas, la tasa de RVS fue menor en los que hicieron lead-in y TGR frente a los que completaron hasta semana 48 (81% vs 96%), mientras que en el subgrupo sin lead-in, la tasa de RVS fue la misma independientemente de la duración de la terapia.
- Eficacia de Faldaprevir en pacientes no respondedores a tratamiento previo. Los resultados en este subtipo de pacientes fueron publicados en el SILENC-223. Al igual, que en el SILENC-122, se excluyeron a pacientes cirróticos.
Se asignaron aleatoriamente a uno de los tres brazos de tratamiento en proporción 2:1:1: grupo 1) 24 semanas de triple terapia (faldaprevir 240 mg QD, pegINF y RBV) con lead-in con PegINF y RBV, seguido de 24 semanas de biterapia con pegINF y RBV; grupo 2) igual que el anterior pero sin lead-in y el grupo 3) 24 semanas de triple terapia (faldaprevir 240 mg do veces al día (BID), pegINF y RBV con período de lead-in) seguido de 24 semanas con biterapia.
La RVS24 osciló entre 32-50% en los pacientes respondedores parciales y entre el 21-35% en los respondedores nulos .En ambos subgrupos de pacientes las tasas más elevadas de RVS fueron alcanzadas en los pacientes que recibieron faldaprevir 240 mg QD sin lead-in.
En cuanto a la TGR, en este caso los resultados no fueron satisfactorios, observándose una disminución significativa de la respuesta en los pacientes que cesaban el tratamiento en la semana 24 frente a los que continuaron tratamiento hasta la semana 48 (43% vs 72%; p = 0,035).
Por tanto, como era esperable, en este estudio se observa un descenso importante de la respuesta (3550%) con respecto a los resultados obtenidos con faldaprevir en pacientes naïve (72-84%). Otro aspecto, que al igual que en el resto de fármacos no ha sido evaluada es la influencia de la cirrosis en la respuesta.
- Seguridad del tratamiento con Faldaprevir. Los EA más frecuentes asociados al tratamiento con faldaprevir fueron: rash, hiperbilirrubinemia, fotosensibilidad y trastornos gastrointestinales (Tabla 6). La tasa de discontinuación por EA osciló entre 4-23%.
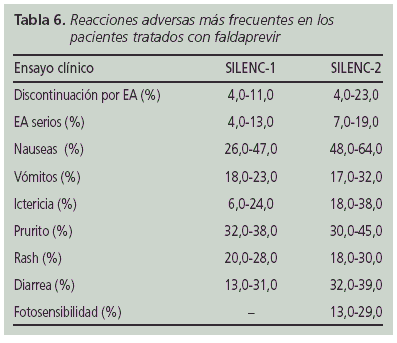
- Variaciones genotípicas asociadas con resistencia. La proporción de pacientes que presentaron repuntes virológicos fueron mayores en el SILENC-223 (17-29%) que en el SILENC-122 (3-6%). En los distintos estudios, en algunos de los pacientes tratados con faldaprevir que presentaron repuntes o fracaso virológico fueron detectadas mutaciones en el dominio de la proteasa NS3 que podían conferir resistencias al fármaco. Éstas mutaciones fueron diferentes en función del genotipo viral, detectándose la R155K en pacientes con genotipo 1a y la D168V en genotipo 1b.
Simeprevir
Simeprevir (TCM435) es un inhibidor de la proteasa NS3/4A26. Este fármaco sólo ha sido evaluado en pacientes infectados por el genotipo 1, en los que como veremos ha mostrado resultados satisfactorios de eficacia y seguridad que han llevado a su reciente aprobación por la FDA. Actualmente disponemos de datos publicados de tres EC en fase II27,28,29.
- Eficacia de Simeprevir en pacientes naïve. El estudio DRAGON27 es un EC en fase IIb, randomizado, abierto y comparativo que incluyó a una cohorte japonesa de pacientes infectados por el genotipo 1b, naïve, y con ausencia de cirrosis. 92 pacientes fueron asignados aleatoriamente a uno de los 5 grupos de tratamiento en proporción 2:1:2:1:1: 12 semanas de triple terapia con simeprevir (SMV) (50 o 100 mg QD ) pegINF y RBV seguido de 12 semanas de biterapia con pegINF y RBV o bien 24 semanas de triple terapia con SMV (50 o 100 mg QD) en combinación con pegINF o RBV o bien 48 semanas con pegINF y RBV. Se realizó TGR, de modo que los pacientes que no presentaron ARN-VHC < 1,4 log 10 UI/mL en la semana 4 y carga viral indetectable en la semana 12, 16 y 20 se mantenía tratamiento con pegINF y RBV hasta la semana 48.
La tasa de RVS24en los grupos tratados con SMV fue del 78-92% comparado con el 46% en el grupo tratado con biterapia. La mayoría de los pacientes tratados con simeprevir presentaron RVR y Respuesta viral temprana completa (RVTc) 83-90 y 92-98%, respectivamente, frente al 8 y 54% en el grupo con biterapia.
El estudio PILLAR28, presenta un diseño similar al anterior, pero este fue controlado con placebo. Al igual que el estudio DRAGON27, excluyó a pacientes cirróticos. Los pacientes fueron asignados aleatoriamente a los 5 grupos de tratamiento que consistieron en 12 o 24 semanas de triple terapia con PegINF/RBV y SMV 75 mg o 150 mg QD o bien 48 semanas de PegINF, RBV y placebo. Al igual que en estudio DRAGON27 se llevó a cabo la TGR en aquellos pacientes que presentaron RVR y RVTc, permitiendo acortar la terapia a 24 semanas. La variable principal de eficacia en este estudio fue la tasa de RVS en la semana 72 independientemente de la duración del tratamiento, aunque también se evaluó la RVS en semanas 12 y 24 post tratamiento.
Un total de 386 pacientes (75-79 por grupo) fueron incluidos en el análisis por intención de tratar. La mayoría de los participantes presentaron una carga viral basal elevada y entre 50-62% fueron genotipo 1b. El 60-78% tenían genotipo no CC para la IL28B.
La tasa de RVS en la semana 72 fue 70,7-84,8% en los pacientes con SMV comparado con el 64,9% en el grupo placebo. Hubo diferencias estadísticamente significativas entre los pacientes que recibieron SMV 150 mg (12 o 24 semanas) y el grupo placebo (77,9-84,8 vs 64,9%). Las tasas de RVS en las semanas 12 y 24 post tratamiento fueron iguales 86,1%. La tasa de RVR en los pacientes tratados con SMV oscilaron entre el 68,0-75.6, de los que 87,9-94,9% alcanzaron RVS24.
No se detectaron diferencias estadísticamente significativas entre la duración de la terapia y la respuesta. Los pacientes con genotipo 1a presentaron tasas de RVS24 menores que los 1b cuando recibieron SMV 75 mg (66,9% y 88,9%, respectivamente), mientras que fueron similares para dosis de SMV 150 mg y para el grupo placebo. La carga viral basal, el grado de fibrosis (F0-2 vs F3), el genotipo (1b vs 1a) y la edad fueron identificados como factores predictores de RVS.
- Eficacia de Simeprevir en pacientes previamente tratados. En octubre de 2013 se publicaron los resultados del estudio ASPIRE29. Este estudio incluyó a pacientes con cirrosis. Se evaluaron distintas regímenes con simeprevir, los grupos de tratamiento 1 y 2, recibieron 12 semanas de triple terapia con PegINF, RBV y simeprevir (100 mg o 150 mg, respectivamente) seguido de 36 semanas de biterapia con PegINF y RBV; los grupos 3 y 4, 24 semanas de triple terapia (con dosis de simeprevir de 100mg o 150 mg) seguido de 36 semanas de biterapia (PegINF y RBV a dosis estándar según peso) y el grupo 5 y 6 recibieron 48 semanas de triple terapia. Los pacientes asignados al grupo placebo recibieron 48 semanas de PegINF, RBV y placebo.
Las tasas de respuesta fueron significativamente superiores en pacientes tratados con SMV comparado con el grupo placebo (61-80% vs 23% ; p < 0,001) y varió según el tipo de respuesta previa: 38%-59% vs 19% en respondedores nulos, en respondedores parciales 48-86% vs9% y en recaedores 77-89% vs 37%. De forma general , los pacientes tratados con SMV 100 mg y 150 mg, las tasas de RVS fueron significativamente superiores con respecto al grupo placebo , 65% (dosis de 100 mg) y 73% (dosis de 150 mg). Sin embargo, la tasa de RVS fue similar independientemente de la duración del tratamiento. Los pacientes con cirrosis presentaron tasas de RVS inferiores (61-62%), aunque a pesar de ello, éstas fueron muy superiores a las alcanzadas en el grupo control (0%).
- Seguridad del tratamiento con simeprevir. La tasa de discontinuación por EA osciló 0-15%. El rash severo y la fotosensibilidad han sido descritas como causas de discontinuación. Las reacciones adversas más frecuentes asociadas al tratamiento con simeprevir fueron rash (incluyendo fotosensibilidad), prurito y náuseas (Tabla 7). También se observó un aumento de los niveles de bilirrubina en los pacientes tratados con simeprevir, auque en la mayoría de los casos fue de grado 1 y reversible cuando se interrumpía el tratamiento (Tabla 7). También han sido detectados algunos casos de disnea en los fase III, aunque esto ha sido poco frecuente.
- Variaciones genotípicas asociadas con resistencia. La tasa de repuntes virológicos en los distintos estudios27,28,29 osciló entre el 4,9%-10,6%. A excepción del estudio DRAGON27, en el que sólo hubo un caso, en el resto de estudios28,29 se detectaron mutaciones en el dominio NS3 que podían conferir resistencia a simeprevir. Por otro lado, la tasa de recidiva, fue del 8-17% en el DRAGON27, del 11,8% en el PILLAR28 y del 10,2% en el ASPIRE29. En la mayoría de estos pacientes se detectaron mutaciones, que a su vez eran diferentes en función del genotipo viral28, siendo la D168X en el genotipo 1b y R155K.
Vaniprevir
Vaniprevir (NK-7009) es otro de los inhibidores de la proteasa NS3/4A de segunda generación.
- Eficacia de vaniprevir en pacientes naïve. Los primeros resultados de eficacia y seguridad se presentaron en un EC en fase 2a30, controlado con placebo, en el que se incluyeron 70 pacientes naïve genotipo 1 a los que se les administró vaniprevir en combinación con PegINF y RBV durante 4 semanas seguido de 44 semanas de biterapia con pegINF y RBV. El objetivo principal de este estudio, además de establecer el rango de dosis de vaniprevir fue determinar la tasa de RVR, aunque también se analizó la tasa de RVS24. En los grupos tratados con distintas dosis de vaniprevir en asociación con biterapia las tasas de RVS oscilaron entre el 61-84%, mientras que en el grupo tratado con placebo, PegINF y RBV fue del 63%, no habiendo, por tanto, diferencias estadísticamente significativas entre ellos. La proporción de pacientes que presentaron RVR si fue significativamente mayor en los grupos tratados con vaniprevir comparado con placebo, 68,8%-83,3% vs 5,6%; p < 0,001).
- Eficacia de vaniprevir en pacientes previamente tratados. La eficacia de la triple terapia con vaniprevir, PegINF alfa 2a y RBV en pacientes con genotipo 1 no respondedores a tratamiento previo con biterapia con pegINF y RBV ha sido evaluada en un EC en fase IIb31. Este estudio excluyó también a pacientes cirróticos. Los pacientes fueron asignados a uno de los 5 grupos de tratamiento: 24 semanas de triple terapia (vaniprevir 600 mg BID, pegINF y RBV); 24 semanas de triple terapia seguido de 24 semanas de pegINF, RBV y placebo, 48 semanas con triple terapia con dosis de vaniprevir de 300 mg BID o 600 mg BID; o bien grupo placebo (48 semanas de pegINF, RBV y placebo). El objetivo principal de este estudio era evaluar la seguridad de las distintas dosis de vaniprevir administradas con biterapia y como objetivos secundarios se estableció la evaluación de la eficacia a través de la RVS24.
En cuanto a las características de los pacientes, podemos destacar que el 80% presentaban genotipo no CC de la IL28B.
Los pacientes que recibieron 24 ó 48 semanas con dosis de vaniprevir de 600 mg BID obtuvieron tasas de
RVS24 del 71% y 78%, respectivamente. Los pacientes asignados a la dosis de 300 mg BID presentaron tasas de RVS24 más bajas (67%), aunque este estudio no fue diseñado para detectar diferencias entre los distintos grupos de tratamiento. El mayor porcentaje de respuesta fue alcanzado en el grupo que recibió 24 semanas de triple terapia con dosis de vaniprevir de 600 mg BID seguido de 24 semanas de pegINF, RBV y placebo (84%). En cualquier caso, las tasas de RVS24 fueron significativamente mayores en todos los grupos tratados con vaniprevir que el grupo placebo, en el que sólo el 19% obtuvieron respuesta (p < 0,001).
- Seguridad del tratamiento con vaniprevir. En relación a la seguridad, los efectos adversos más comunes asociados al tratamiento con vaniprevir fueron de tipo gastrointestinal (diarrea, náuseas y vómitos). En cuanto a la tasa de discontinuación por efectos adversos, en el estudio llevado a cabo en pacientes naïve ningún paciente discontinuó el tratamiento, mientras que en el otro estudio la tasa de discontinuación fue del 11%.
- Variaciones genotípicas asociadas con resistencia. En ambos estudios se han detectado mutaciones en las posiciones R155 y D168 de NS3 que han sido asociadas con resistencia al tratamiento con vaniprevir en pacientes que presentaron fracaso virológico30,31.
Combinaciones de AAD
Daclatasvir y asunaprevir
Daclatasvir ha sido evaluado tanto en combinación con pegINF y RBV, como asociado asunaprevir (BMS-650032), otro AAD inhibidor de la proteasa NS332.
- Eficacia de asunaprevir y daclatasvir en pacientes previamente tratados. El primer estudio fue publicado en el año 201132. Se trata de un EC en fase IIa, abierto y randomizado, que incluyó 21 pacientes genotipo 1, no respondedores a tratamiento previo con biterapia y con ausencia de cirrosis. Los regímenes comparados la combinación de daclatasvir y asunaprevir (grupo A) con asunaprevir y daclatasvir en combinación con pegINF y RBV (grupo B) administrados ambos durante 24 semanas. Las dosis de daclatasvir y asunaprevir empleadas fueron 60 mg QD y 600 mg BID, respectivamente. Los pacientes del grupo A que presentaron repunte virológico se les añadió PegINF y RBV y se mantuvo hasta la semana 48.
La mayoría fueron genotipo 1a y presentaban genotipo CT o TT de la IL28B. La tasa de RVS12 fue del 100% de los pacientes del grupo B (daclatasvir y asunaprevir en combinación con pegINF y RBV) frente al 64% en el grupo A.
En otro estudio llevado a cabo por Suzuky y colaboradoresBB en una cohorte de pacientes japonenes, se evaluó la biterapia con asunaprevir (200 mg BID) y daclatasvir (60 mg QD) en pacientes respondedores nulos o bien intolerantes o incompatibles a recibir tratamiento con PegINF y RBV. En este estudio la tasa de RVS12 global en la cohorte fue del 77% y del 90% en los respondedores nulos y del 67% en los intolerantes/incompatibles a PegINF y RBV.
En cuanto a las características de los pacientes, aproximadamente la mitad de los pacientes presentaban genotipo CC de la IL28B. Ninguna de las características basales (genotipo IL28B, grado de fibrosis o la carga viral basal) fue asociada con menores tasas de RVS.
- Seguridad del tratamiento con asunaprevir y daclatasvir. Los efectos adversos más frecuentes asociados a la combinación de estos fármacos en los distintos estudios fueron diarrea, dolor de cabeza, náuseas y elevación transitoria de las transaminasas. Éste último ha sido asociado principalmente a asunaprevir. La tasa de discontinuación por EA fue variable 0-7% (Tabla 8).
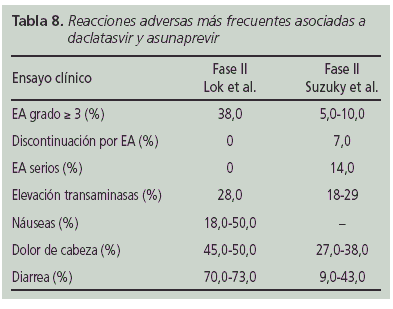
- Variaciones genotipicas asociadas con resistencia. En ambos estudiosB2,BB han sido detectados polimorfismos asociados con resistencias a daclatasvir y asunaprevir, algunos de ellos no han tenido repercusión en la respuesta, mientras que otros han sido detectados en pacientes que han presentado fracaso virológico.
Daclatasvir, asunaprevir y BMS-791325
- Eficacia de la combinación de tres AAD. BMS-791B25 es otro inhibidor de la polimerasa NS5B. La combinación de estos tres AAD ha sido evaluada en un EC en fase IIB4 con el objetivo de conocer si la adición de un tercer AAD puede mejorar las tasas de RVS en pacientes con genotipo 1, sin la necesidad de asociar pegINF y RBV. Este estudio incluyó a pacientes naïve , genotipo 1, no cirróticos. Los brazos de tratamiento fueron: los grupos 1 y 2 recibieron 24 o 12 semanas de triple terapia con dosis de BMS- 791B25 de 75 mg BID y los grupos B y 4 recibieron 24 o 12 semanas de triple terapia con dosis de BMS-791B25 de 150 mg BID. La dosis de daclatasvir empleada fue de 60 mg QD y asunaprevir 200 mg BID.
En cuanto a las características de los pacientes, destacar que el 64% de los pacientes eran genotipo 1a y el 70% tenían genotipo no CC de la IL28B y más del 50% presentaban F2 o mayor, aunque ninguno de ellos se comportó como un factor predictivo de RVS. El 94% de los pacientes asignados a los grupos 1 y 2 alcanzaron RVS12, mientras que en el 94% y 89% en los grupos B y 4, respectivamente. De los 66 pacientes, sólo uno presentó recaída post tratamiento y hubo dos casos de repuntes virológicos.
- Seguridad del tratamiento. En cuanto a la seguridad, los efectos adversos más frecuentes fueron: dolor de cabeza, astenia, diarrea, náuseas y dolor abdominal. No hubo ningún caso de discontinuación relacionado con la toxicidad del tratamiento.
- Variaciones genotipicas asociadas con resistencia. En los pacientes con fracaso virológico a la triple terapia fueron detectados polimorfismos en las polimerasas NS5A, NS5B y NSB que podrían conferir resistencias a éstos fármacos.
Sofosbuvir y daclatasvir
- Eficacia. Los datos de eficacia un EC en fase II, abierto y aleatorizadoB5. Este estudio incluyó pacientes naïve con genotipos 1, 2 y B y también una pequeña cohorte de pacientes con genotipo 1 con fracaso virológico al tratamiento con triple terapia con boceprevir y telaprevir. Se excluyeron a pacientes cirróticos en este estudio. Los pacientes con genotipos 2 y B (n = 15B) fueron aleatorizados (en proporción 1:1:1) a recibir una semana de sofosbuvir seguido de 2B semanas de sofosbuvir y daclatasvir o a 24 semanas de sofosbuvir y daclatasvir con o sin RBV. Los pacientes con genotipo 1 (n = 126) fueron aleatorizados a los mismos regímenes de tratamiento y a 12 semanas de tratamiento de sofosbuvir y daclatasvir con o sin RBV. Por otro lado, aquellos con genotipo 1 con fracaso virológico a triple terapia (n = 41) fueron aleatorizados a 24 semanas de sofosbuvir y daclatasvir con o sin RBV. Sofosbuvir y daclatasvir fueron administrados a las dosis de 400 mg QD y 60 mg QD, respectivamente. La dosis de RBV fue de 1000 o 1200 mg al día según el peso en los pacientes con genotipo 1 y de 800 mg al día en los genotipos 2 y 3. La variable principal de eficacia fue la RVS12.
La tasa de RVS12 en los pacientes con genotipos 2 y 3 fue del 91% (92% en genotipo 2 y 89% en genotipo 3) y del 98% en los pacientes con genotipo 1. La tasa de RVS en pacientes con genotipo 1 naïve y no respondedores fue del 98% en ambos grupos. En cuanto a las tasas de RVS en función de los distintos regímenes administrados también fueron similares. Sólo hubo un caso de recaída post tratamiento en un paciente con genotipo 3 cuyo régimen era libre de RBV.
La tasa de RVS en los pacientes con genotipo 1 fue similar en los distintos subgrupos de pacientes: subtipo viral (genotipo 1b vs 1b), genotipo IL28B, raza, régimen con o sin RBV , o si el paciente era naïve o no respondedor a tratamiento con triple terapia.
- Seguridad. En cuanto a la seguridad del tratamiento, los efectos adversos más frecuentes fueron fatiga, náuseas y vómitos. El aumento de los niveles de fosfato y glucemia en sangre fueron las anormalidades de laboratorio grado 3 y 4 más frecuentes.
- Variaciones genotípicas asociadas con resistencia. En varios pacientes fue detectado el polimorfismo NS5A-A30K asociado a resistencia a daclatasvir, sin embargo, sólo un paciente con genotipo 3 en el que se detectó este polimorfismo presentó recaída post tratamiento.
Sofosbuvir y ledipasvir
Otra de las combinaciones evaluadas ha sido las asociación de sofosbuvir con ledipasvir (GS-5885), otro inhibidor de la proteasa NS5A. Este fármaco ha mostrado alta actividad antiviral frente a los genotipos 1a y 1b y además presenta actividad en presencia de la mutación S282T, la única variante hasta ahora conocida que reduce la respuesta a sofosbuvir36. Ledipasvir, al igual que daclatasvir, también inhibidor de la polimerasa NS5A, a pesar de su potente actividad antiviral, requieren ser administrados en combinación con otros AAD para evitar la aparición de mutaciones que ocasionen resistencias al tratamiento.
Análisis de la eficacia
En febrero de 2014 han sido publicados los resultados del estudio LONESTAR37, un EC en fase II, abierto y aleatorizado, en el que se incluyeron 100 pacientes con genotipo 1. 60 pacientes naïve no cirróticos fueron aleatorizados a recibir sofosbuvir y ledipasvir durante 8 semanas con o sin RBV, o a 12 semanas de tratamiento con sofosbuvir y ledipasvir. Por otro lado, 40 pacientes no respondedores a tratamiento previo con triple terapia con boceprevir o telaprevir fueron asignados aleatoriamente a recibir la combinación de los dos AAD durante 12 semanas con o sin RBV. Sofosbuvir y ledipasvir fueron administrados en un solo comprimido que contenía 400 mg y 90 mg, respectivamente. La variable principal de eficacia fue la RVS12.
En cuanto a las características basales de los pacientes destacar que la mayoría de los pacientes tanto naïve como no respondedores presentaban genotipo no CC de la IL28B, 80% Y 93%, respectivamente. Por otro lado, el 55% de los pacientes no respondedores presentaban cirrosis.
La tasa de RVS12 fueron similares en las dos cohortes de tratamiento. El 95% de los pacientes naïve que recibieron la terapia sin RBV independientemente de la duración (8 o 12 semanas) presentaron RVS12 y el 100% cuando se administró con RBV. La misma situación ocurrió con la cohorte de no respondedores, el 95% de los que recibieron terapia sin RBV y el 100% con RBV. Por tanto, la mayoría de los pacientes respondieron a la terapia independientemente de la presencia o ausencia de RBV, la presencia o ausencia de cirrosis o la raza.
- Seguridad. En cuanto a la seguridad, las reacciones adversas fueron más frecuentes en los grupos que recibieron tratamiento con RBV. Los efectos adversos más comunes fueron náuseas, anemia, infecciones del tracto respiratorio superior y dolor de cabeza. Ningún paciente discontinuó el tratamiento por motivos de seguridad.
- Variaciones genotípicas asociadas con resistencia. De los 100 pacientes incluidos en el estudio, dos presentaron recaída post tratamiento. Los dos recibieron la terapia sin RBV y además fueron detectadas variantes asociadas con resistencia a ledipasvir, y también en uno de ellos se detectó la mutación S282T que reduce la eficacia de sofosbuvir.
Faldaprevir y deleobuvir
- Eficacia. Deleobuvir (BI207127) es un inhibidor no nucleósido de la polimerasa NS5B. La actividad de este AAD se ha evaluado en EC en fase I en combinación con faldaprevir y RBV durante cuatro semanas con resultados prometedores38. Recientemente han sido publicados los resultados del estudio SOUND-C239 en el que se analiza la eficacia de la combinación de estos dos AAD con o sin RBV en pacientes naïve con genotipo 1. En este estudio se evaluaron distintos regímenes de tratamiento: 16, 28 o 40 de semanas de triple terapia con faldaprevir (120 mg QD), deleobuvir [600 mg tres veces al día (TID)] y RBV a dosis estándar según peso (grupos TID16, TID28 Y TID40, respectivamente); 28 semanas de triple terapia con faldaprevir (120 mg QD), deleobuvir (600 mg BID) y RBV (grupo BID28) o bien 28 semanas de biterapia con faldaprevir (120 mg QD) y deleobuvir (600 mg TID) y sin RBV (grupo TID28-NR). La variable principal de eficacia fue la RVS12.
En cuanto a las características basales, podemos destacar que el 9% de los pacientes eran cirróticos y el 74% de los pacientes presentaban genotipo no CC de la IL28B.
La tasa de RVS12 en los distintos subgrupos de tratamiento osciló entre el 39% y el 69%. No hubo diferencias estadísticamente significativas en la respuesta en función de la duración del tratamiento (RVS12 del 59%, 59% y 52% en los grupos TID16, TID28 Y TID40, respectivamente) ni en la dosis de deleobuvir administrada (69% en el grupo BID28 vs 59% en el grupo TID28). Sin embargo, si que hubo diferencias significativas en la respuesta a favor de los pacientes que recibieron el régimen con o sin RBV (59% en el gupo TID28 frente al 39% en el grupo TID28-NR, p = 0,03).
En este estudio además se identificaron algunos factores predictores de RVS12, como fueron: sexo femenino, genotipo 1b, genotipo CC IL28B, y regímenes que contuvieran RBV.
- Seguridad. En cuanto a la seguridad, los efectos adversos más frecuentes fueron náuseas, diarrea, vómitos, ictericia, prurito, rash, reacciones de fotonsensibilidad, sequedad de piel, astenia y fatiga. Un 7% de los pacientes desarrollaron reacciones de fotosensibilidad graves. La tasa de discontinuación por efectos adversos fue del 12%.
- Variaciones genotípicas asociadas con resistencia. El 21% de los pacientes presentaron repuntes virológicos, de los que en el 97% de los casos habían desarrollado mutaciones tanto en NS3 como NS5B. El 6% tuvieron recaída tras finalizar el tratamiento.
ABT-450/r y ABT-333
- Eficacia. En un EC en fase II40 ha sido evaluada la combinación de ABT-450, un inhibidor de la proteasa NS3 potenciado con dosis bajas de ritonavir (ABT-450/r) con ABT-333, un inhibidor no nucleósido de la polimerasa NS5B y RBV durante 12 semanas. Esta combinación fue evaluada en 50 pacientes infectados por el genotipo 1, no cirróticos, tanto naïve (n = 33) como respondedores parciales y nulos a biterapia con pegINF y RBV (n = 17). El objetivo principal de este estudio fue evaluar la tasa de RVRe, aunque también se determinó la tasa de RVS12.
La tasa de RVS12 en los pacientes naïve fue del 94%, mientras que en los no respondedores fue del 47%.
En este estudio es destacable que el 88% de los pacientes presentaban genotipo 1a y el 70% genotipo no CC IL28B. Sin embargo, ninguno de estos factores fue asociado con la respuesta.
- Seguridad. En cuanto a la seguridad, el tratamiento fue bien tolerado y sólo un paciente tuvo que discontinuar el tratamiento. Los efectos adversos más frecuentes fueron fatiga, nauseas, dolor de cabeza, discinesia insomnio, prurito, rash y vómitos.
- Variaciones genotípicas asociadas con resistencia. En 8 de los 9 pacientes que presentaron fracaso virológico fueron detectadas variantes genotípicas en NS3 Y NS5B.
ABT-450/r y ABT-072
Otro EC también en fase IIa41 evalúa de nuevo ABT-450/r combinado con otra molécula (ABT-072), un inhibidor de la polimerasa NS5B y con RBV. Este estudio fue llevado a cabo en 11 pacientes naïve, genotipo 1, no cirróticos y con genotipo CC de la IL28B. Ambos AAD fueron administrados en una sola toma diaria en combinación con dosis estándar de RBV durante 12 semanas. La tasa de RVS24 fue del 91%, aunque un paciente recayó en la semana 36 post tratamiento. En cuanto a la seguridad, no hubo discontinuaciones por efectos adversos y los más frecuentes observados fueron: dolor de cabeza, náuseas, fatiga y sequedad de piel.
Discusión
La incorporación de estos nuevos AAD van a permitir un avance muy importante en el tratamiento de la hepatitis C, alcanzando tasas de erradicación del virus cercanas al 100%. Ya la aparición de la primera generación de IP (BOC y TVR) supuso un cambio radical en el escenario terapéutico de la enfermedad, gracias al aumento drástico en las tasas de RVS comparadas con las alcanzadas con la terapia estándar basada en PegINF y RBV. Sin embargo, se trata de regímenes terapéuticos muy complejos y con un perfil importante de efectos secundarios, siendo algunos de ellos de carácter grave42. Por ello, el conocimiento en mayor profundidad del proceso de replicación del virus ha permitido el desarrollo de nuevas moléculas que optimicen la eficacia y la seguridad y que además disminuyan la complejidad de la terapia.
Los inhibidores de la proteasa NS5A constituye una de las nuevas familias de AAD, siendo daclatasvir la primera molécula desarrollada de esta familia. Este fármaco ha sido el único evaluado como único IP administrado con PegINF y RBV, cuyos resultados no han sido muy satisfactorios12, fundamentalmente ocasionado por la mayor probabilidad que tiene esta familia de IP de desarrollar resistencias. Esto ha llevado a que el resto de moléculas de esta familia sean combinadas con otros IP con el fin de mejorar las tasas de RVS.
Por el contrario, sofosbuvir, uno de los inhibidores de la polimerasa NS5B, ha mostrado una barrera genética alta, permitiendo por primera vez la aprobación de un fármaco frente al VHC con regímenes libres de INF. En la única circunstancia en la que se detectaron mutaciones que podían conferir resistencias fue en el estudio ELECTRON15 asociadas al uso de sofosbuvir en monoterapia. Otro de los aspectos diferenciales de sofosbuvir es que presenta actividad en todos los genotipos virales. A pesar de aproximadamente el 80% de los pacientes con genotipos 2 y 3 responden a biterapia con PegNF y RBV, hasta ahora no existían opciones terapéuticas para aquellos pacientes que fracasan al tratamiento estándar con biterapia o bien que no toleran o no pueden recibir tratamiento con INF. Las tasas de RVS con sofosbuvir y RBV con o sin PegINF en pacientes naïve oscilan en torno al 90%15,16,18 ,mientras que en los previamente tratados no llegan al 80%20.
En cuanto a la segunda generación de inhibidores de la proteasa NS3/4A, simeprevir ha sido el primero aprobado por la FDA, alcanzando respuestas próximas al 90% en pacientes naïve27,28, mientras que en pacientes previamente tratados con PegINF/RBV éstas no superaban el 80%29. Por el contrario, la actividad de faldaprevir ha sido muy superior en pacientes naïve comparada con aquellos no respondedores a biterapia23. Vaniprevir, ha mostrado tasas de respuesta similares en pacientes naïve y pretratados3031. Sin embargo, uno de los aspectos negativos de esta familia de fármacos, es que requieren su administración con PegINF/RBV.
El riesgo en la aparición de resistencias de la mayoría de los IP dificulta la posibilidad de su administración en regímenes libres de pegINF y RBV e incluso su administración como único AAD. Por ello, se han ido realizando distintos estudios en los que se evalúan la combinaciones de dos e incluso tres AAD con o sin la presencia de RBV. Entre ellas, la combinación de asunaprevir y daclatasvir parecen ser prometedoras con resultados favorables en pacientes no respondedores a tratamiento con biterapia32,33, así como la asociación de estos dos fármacos con BMS-791325 en pacientes naïve34. Sin embargo, las dos combinaciones a priori con resultados más impactantes son las de sofosbuvir y daclatasvir35 y sofosbuvir y ledipasvir37. Ambos regímenes en presencia o ausencia de RBV muestran tasas de RVS similares y como aspecto más importante es que se alcanzan tasas de RVS próximas al 100% en pacientes no respondedores a triple terapia con BOC o TVR. Sin embargo, es necesario confirmar estos resultados en estudios con un mayor tamaño muestral. Además la asociación de sofosbuvir y daclatasvir también mostró altas tasas de RVS en pacientes con genotipos 2 y 3. Por el contrario, otras combinaciones como faldaprevir y deleobuvir no han obtenido resultados tan favorables39.
En cuanto a las características de los pacientes incluidos en los ensayos, es destacable que la mayoría de los estudios, excluían a pacientes cirróticos, o bien constituían un mínimo porcentaje de la población incluida en el ensayo. La presencia de cirrosis constituye una de las principales variables asociadas con menores tasas de RVS. En varios estudios con sofosbuvir18,20, así como con simeprevir28,29 ha sido asociada de forma significativa con menores respuestas. Por otro lado, algunos fármacos no incluyen en ninguno de sus estudios a pacientes con cirrosis, como es el caso de daclatasvir, faldaprevir y vaniprevir, así como en la mayoría de las asociaciones de AAD evaluadas. Resultados preliminares de un EC en fase III con faldaprevir revelan que la ausencia de cirrosis constituye un factor predictivo de RVS25. Por el contrario, según los resultados preliminares de un EC en pacientes tratados con asunaprevir y daclatasvir, la respuesta fue similar independientemente de la presencia o no de cirrosis43.
En cuanto al subtipo viral, en algunos estudios, el genotipo 1b ha sido identificado como un factor predictivo de RVS28,34,39. Esto podría estar relacionado con la mayor probabilidad de desarrollo de resistencias en los pacientes con genotipo 1a.
En cuanto a la influencia del genotipo de la IL28B en la respuesta a los nuevos IP no está claro, ya que los resultados son variables. De todos los estudios llevados a cabo con sofosbuvir sólo uno de los EC en fase III18, el genotipo CC fue identificado como un factor predictivo de RVS, mientras que en los demás estudios no. En el resto de fármacos analizados en esta revisión, sólo las combinaciones de daclatasvir, asunaprevir y BMS-79132534 y de faldaprevir y deleobuvir39 identificaron el genotipo de la IL28B como un factor asociado con la respuesta.
Por otro lado, en la mayoría de los fármacos han sido detectadas variaciones genotípicas asociadas con resistencias. Algunas familias, como los inhibidores de proteasa NS5A tienen mayor susceptibilidad al desarrollo de resistencias, sin embargo, con la combinación con otros AAD pueden ser disminuidas de forma sustancial o bien no tener impacto en la respuesta.
Finalmente, en el ámbito de la seguridad, es más que evidente que éstas nuevas generaciones de AAD son mucho mejor toleradas, con tasas de discontinuación por motivos de seguridad bajas. En muchos casos, el perfil de seguridad obviamente se ve favorecido por la ausencia en muchos de los regímenes de la presencia de PegINF y de RBV.
Por lo tanto, una vez que esta nueva generación de fármacos se autoricen en nuestro país será necesario diseñar estrategias y las líneas prioritarias de actuación que nos dirijan en la toma de decisiones y selección de los pacientes candidatos a cada una de las diferentes opciones terapéuticas próximamente disponibles, así como saber en que momento tratar, ya que la cirrosis constituye uno de los principales problemas que pueden limitar la eficacia de estos nuevos fármacos.
Conclusiones
La aparición en el mercado de las nuevas generaciones de inhibidores de la proteasa frente al VHC van a suponer un aumento de las tasas de curación en todos los subtipos de pacientes, a través de regímenes más sencillos y mejor tolerados. El nuevo arsenal terapéutico disponible frente a esta enfermedad nos obliga a identificar a aquellos pacientes que más se van a beneficiar de cada una de las estrategias disponibles.
Bibliografía
1. Poynard T, Yuen M-F, Ratziu V, Lai CL. Viral hepatitis C. Lancet. 2003;362(9401):2095-100. [ Links ]
2. Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958-65. [ Links ]
3. Thomas DL. Advances in the treatment of hepatitis C virus infection. Top Antivir Med. 2012;20(1):5-10. [ Links ]
4. AEMPS - Centro de Información online de Medicamentos de la AEMPS (CIMA) - Buscador principal (Internet). (cited 2013 Dic 10). Available from: http://www.aemps.gob.es/cima/fichasTecnicas.do?metodo=buscar. [ Links ]
5. US Food and Drug Administration Home Page (Internet). (cited 2013 Dic 10). Available from: http://www.fda.gov/. [ Links ]
6. Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1207-17. [ Links ]
7. Poordad F, McCone J Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1195-206. [ Links ]
8. Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364(25):2405-16. [ Links ]
9. McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med. 2009;361(6):580-93. [ Links ]
10. Chopra A, Klein PL, Drinnan T, Lee SS. How to optimize HCV therapy in genotype 1 patients: management of side-effects. Liver Int Off J Int Assoc Study Liver. 2013;33(Suppl. 1):30-4. [ Links ]
11. Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465(7294):96-100. [ Links ]
12. Pol S, Ghalib RH, Rustgi VK, Martorell C, Everson GT, Tatum HA et al. Daclatasvir for previously untreated chronic hepatitis C genotype-1 infection: a randomised, parallel-group, double-blind, placebo-controlled, dose-finding, phase 2a trial. Lancet Infect Dis. 2012;12(9):671-7. [ Links ]
13. Lam AM, Murakami E, Espiritu C, Steuer HMM, Niu C, Keilman M et al. PSI-7851, a pronucleotide of beta-D-2'-deoxy-2'-fluoro-2'-C-methyluridine monophosphate, is a potent and pan-genotype inhibitor of hepatitis C virus replication. Antimicrob Agents Chemother. 2010;54(8):3187-96. [ Links ]
14. Label information Sofosbuvir. FDA. 2013. (citado 10/12/2013). Disponible en: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204671s000lbl.pdf. [ Links ]
15. Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013;368(1):34-44. [ Links ]
16. Lawitz E, Lalezari JP, Hassanein T, Kowdley KV, Poordad FF, Sheikh AM et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naivfe patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis. 2013;13(5):401-8. [ Links ]
17. Kowdley KV, Lawitz E, Crespo I, Hassanein T, Davis MN, DeMicco M et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet. 2013;381(9883):2100-7. [ Links ]
18. Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368(20):1878-87. [ Links ]
19. Osinusi A, Meissner EG, Lee Y-J, Bon D, Heytens L, Nelson A, et al. Sofosbuvir and ribavirin for hepatitis C genotype 1 in patients with unfavorable treatment characteristics: a randomized clinical trial. JAMA J Am Med Assoc. 2013;310(8):804-11. [ Links ]
20. Jacobson IM, Gordon SC, Kowdley KV, Yoshida EM, Rodriguez-Torres M, Sulkowski MS et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med. 2013;368(20):1867-77. [ Links ]
21. Manns MP, Bourliére M, Benhamou Y, Pol S, Bonacini M, Trepo C et al. Potency, safety, and pharmacokinetics of the NS3/4A protease inhibitor BI201335 in patients with chronic HCV genotype-1 infection. J Hepatol. 2011;54(6):1114-22. [ Links ]
22. Sulkowski MS, Asselah T, Lalezari J, Ferenci P, Fainboim H, Leggett B et al. Faldaprevir combined with pegylated interferon alfa-2a and ribavirin in treatment-naTve patients with chronic genotype 1 HCV: SILEN-C1 trial. Hepatol Baltim Md. 2013;57(6):2143-54. [ Links ]
23. Sulkowski MS, Bourliére M, Bronowicki J-P, Asselah T, Pawlotsky J-M, Shafran SD et al. Faldaprevir combined with peginterferon alfa-2a and ribavirin in chronic hepatitis C virus genotype-1 patients with prior nonresponse: SILEN-C2 trial. Hepatol Baltim Md. 2013;57(6):2155-63. [ Links ]
24. Nishiguchi S, Sakai Y, Kuboki M, Tsunematsu S, Urano Y, Sakamoto W et al. Safety and efficacy of faldaprevir with pegylated interferon alfa-2a and ribavirin in Japanese patients with chronic genotype-1 hepatitis C infection. Liver Int Off J Int Assoc Study Liver. 2014;34(1):78-88. [ Links ]
25. HIVandHEPATITIS.com. San Francisco. (cited 2013 Dic 10). Available from: http://hivandhepatitis.com/hcv-treatment/experimental-hcvdrugs/4429-aasld-2013-faldaprevir-triple-therapy-effective-acrosspatient-subgroups. [ Links ]
26. Manns M, Reesink H, Berg T, Dusheiko G, Flisiak R, Marcellin P et al. Rapid viral response of once-daily TMC435 plus pegylated interferon/ribavirin in hepatitis C genotype-1 patients: a randomized trial. Antivir Ther. 2011;16(7):1021-33. [ Links ]
27. Hayashi N, Seto C, Kato M, Komada Y, Goto S. Once-daily simeprevir (TMC435) with peginterferon/ribavirin for treatment-naTve hepatitis C genotype 1-infected patients in Japan: the DRAGON study. J Gastroenterol. 2013. [ Links ]
28. Fried MW, Buti M, Dore GJ, Flisiak R, Ferenci P, Jacobson I et al. Once-daily simeprevir (TMC435) with pegylated interferon and ribavirin in treatment-naTve genotype 1 hepatitis C: The randomized PILLAR study. Hepatol Baltim Md. 2013;58(6):1918-29. [ Links ]
29. Zeuzem S, Berg T, Gane E, Ferenci P, Foster GR, Fried MW et al. Simeprevir Increases Rate of Sustained Virologic Response Among Treatment-Experienced Patients With HCV Genotype-1 Infection: A Phase IIb Trial. Gastroenterology. 2013 Nov 1. [ Links ]
30. Manns MP, Gane E, Rodriguez-Torres M, Stoehr A, Yeh C-T, Marcellin P et al. Vaniprevir with pegylated interferon alpha-2a and ribavirin in treatment-naTve patients with chronic hepatitis C: a randomized phase II study. Hepatol Baltim Md. 2012;56(3):884-93. [ Links ]
31. Lawitz E, Rodriguez-Torres M, Stoehr A, Gane EJ, Serfaty L, Bhanja S et al. A phase 2B study of MK-7009 (vaniprevir) in patients with genotype 1 HCV infection who have failed previous pegylated interferon and ribavirin treatment. J Hepatol. 2013;59(1):11-7. [ Links ]
32. Lok AS, Gardiner DF, Lawitz E, Martorell C, Everson GT, Ghalib R et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med. 2012;366(3):216-24. [ Links ]
33. Suzuki Y, Ikeda K, Suzuki F, Toyota J, Karino Y, Chayama K et al. Dual oral therapy with daclatasvir and asunaprevir for patients with HCV genotype 1b infection and limited treatment options. J Hepatol. 2013;58(4):655-62. [ Links ]
34. Everson GT, Sims KD, Rodriguez-Torres M, Hézode C, Lawitz E, Bourlière M, et al. Efficacy of an interferon- and ribavirin-free regimen of daclatasvir, asunaprevir, and BMS-791325 in treatment-naive patients with HCV genotype 1 infection. Gastroenterology. 2014;146(2):420-9. [ Links ]
35. Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, Jacobson I et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 2014;370(3):211-21. [ Links ]
36. Nakamoto S, Kanda T, Wu S, Shirasawa H, Yokosuka O. Hepatitis C virus NS5A inhibitors and drug resistance mutations. World J Gastroenterol WJG. 2014;20(11):2902-12. [ Links ]
37. Lawitz E, Poordad FF, Pang PS, Hyland RH, Ding X, Mo H, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet. 2014;383(9916):515-23. [ Links ]
38. Zeuzem S, Asselah T, Angus P, Zarski J-P, Larrey D, Müllhaupt B et al. Faldaprevir (BI 201335), deleobuvir (BI 207127) and ribavirin oral therapy for treatment-naive HCV genotype 1: SOUND-C1 final results. Antivir Ther. 2013;18(8):1015-9. [ Links ]
39. Zeuzem S, Soriano V, Asselah T, Bronowicki J-P, Lohse AW, Müllhaupt B et al. Faldaprevir and deleobuvir for HCV genotype 1 infection. N Engl J Med. 2013;369(7):630-9. [ Links ]
40. Poordad F, Lawitz E, Kowdley KV, Cohen DE, Podsadecki T, Siggelkow S et al. Exploratory study of oral combination antiviral therapy for hepatitis C. N Engl J Med. 2013;368(1):45-53. [ Links ]
41. Lawitz E, Poordad F, Kowdley KV, Cohen DE, Podsadecki T, Siggelkow S et al. A phase 2a trial of 12-week interferon-free therapy with two direct-acting antivirals (ABT-450/r, ABT-072) and ribavirin In IL28B C/C patients with chronic hepatitis C genotype 1. J Hepatol. 2013;59(1):18-23. [ Links ]
42. Hézode C. Boceprevir and telaprevir for the treatment of chronic hepatitis C: safety management in clinical practice. Liver Int Off J Int Assoc Study Liver. 2012;32(Suppl. 1):32-8. [ Links ]
43. Chayama K, Suzuki Y, Ikeda K, Toyota J, Karino Y, Kawasaki Y et al. All-oral Combination of Daclatasvir Plus Asunaprevir in Interferon Ineligible Naive/Intolerant and Nonresponder Japanese Patients Chronically Infected with HCV Genotype 1b: Results from a Phase 3 Trial. 64th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD 2013). Washington, DC, November 1-5, 2013. Abstract211. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Correo electrónico: rociojg85@hotmail.com
(Rocío Jiménez Galán).
Recibido el 31 de enero de 2014
Aceptado el 4 de abril de 2014

