










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkFarmacia Hospitalaria
versión On-line ISSN 2171-8695versión impresa ISSN 1130-6343
Farm Hosp. vol.44 no.6 Toledo nov./dic. 2020 Epub 27-Dic-2021
https://dx.doi.org/10.7399/fh.11353
ORIGINALES
Desarrollo y validación de un panel de secuenciación masiva en paralelo para farmacogenética clínica
1Health in Code S. L., Science Department, A Coruña. Spain.
2 Biomedical Research Institute, A Coruña. Spain.
3 Universidade da Coruña, GRINCAR (Cardiovascular Research Group), A Coruña. Spain.
Introducción
La farmacogenética es la disciplina que estudia la influencia del genotipo en la respuesta a los medicamentos. La farmacogenética clínica propone estrategias individualizadas para el manejo de los medicamentos en base al genotipo de cada paciente. El fin último de la farmacogenética es la mejora de los resultados en salud de los tratamientos farmacológicos, eficacia y efectos adversos, así como la mejora en relación coste-beneficio; por tanto, se trata de una estrategia para el uso racional del medicamento1.
La incorporación de los datos genéticos en los procesos asistenciales debe sustentarse en una buena tecnología de secuenciación de alta calidad, así como en procesos de análisis e interpretación de resultados estandarizados. El desarrollo en los últimos años de las técnicas de secuenciación masiva en paralelo o NGS (del inglés: next-generation sequencing) ha supuesto un salto tecnológico disruptivo. Se ha pasado de la capacidad de analizar decenas de variantes a la posibilidad de estudiar decenas de miles de variantes en un único estudio. Su incorporación a los procesos de diagnóstico médico es cada vez más frecuente; sin embargo, en el ámbito de la farmacogenética se continúa empleando tecnologías convencionales como reacción en cadena de la polimerasa cuantitativa, métodos de extensión de primer tales como TaqMan® (Thermofisher®) o MassARRAY® (Sequenom®)2,3.
El empleo de estas tecnologías convencionales no permite un estudio en profundidad de todas las regiones genéticas de interés y obliga a estudiar solamente un subconjunto de ellas. Diversas revisiones y estudios comparativos enfocados a las técnicas de farmacogenética han puesto de manifiesto que existen discrepancias en el análisis e interpretación de los resultados entre los diferentes laboratorios y tecnologías. Estas discrepancias se han atribuido a que los diversos laboratorios analizan regiones genéticas diferentes sin incluir todo el espectro de variantes de interés4-6. Todo ello, des graciadamente, reduce el valor clínico de los estudios, motiva la desconfianza en los resultados, dificultando y retrasando la incorporación de una farmacogenética de más valor en la práctica clínica asistencial.
En este artículo se expone el diseño, desarrollo, implementación y validación de un panel de secuenciación NGS de farmacogenética orientado a la práctica clínica.
Métodos
Definición de las regiones genéticas de interés clínico
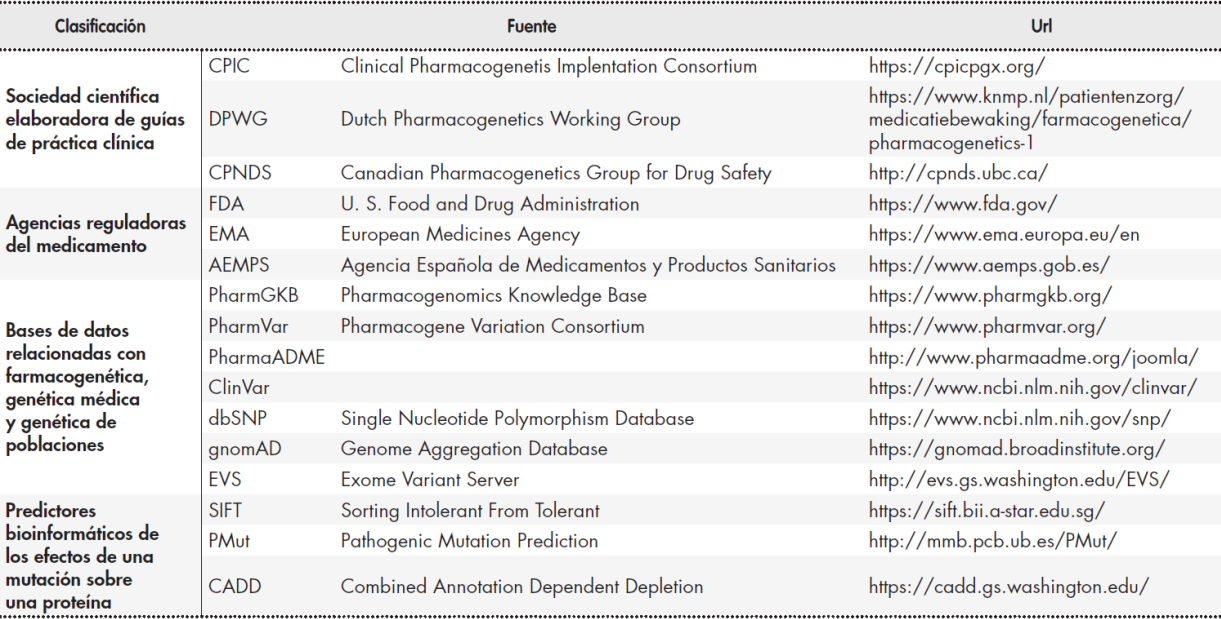
La selección de las regiones de interés se realizó mediante búsqueda bibliográfica. Una región se consideró clínicamente relevante si su análisis aporta información que pueda modificar la estrategia terapéutica asistencial para un fármaco (por ejemplo, selección del tratamiento, dosificación, seguimiento del paciente). Las fuentes bibliográficas principales empleadas comprenden guías de práctica clínica de sociedades científicas farmacogenéticas, fichas técnicas de agencias reguladoras del medicamento y bases de datos relacionadas con la genética que se detallan en la Tabla 1. Las principales plataformas de farmacogenética convencional analizadas fueron Affymetrix DMET® y Agena Bioscience iPLEX®. Ciertas regiones genéticas como CYP2D6 o HLA-B requirieron de un diseño específico. Para CYP2D6 se diseñaron sondas para la captura de todas las regiones codificantes y para las regiones de alta homología (CYP2D7 y CYP2D8) con el objeto de estudiar variaciones en el número de copias o CNV (del inglés: copy number variation) y variantes estructurales. Para HLA-B se realizó un diseño de sondas frente a las secuencias de referencia de la base de datos IMGT/HLA7 similar a la aproximación de Wittig et al.8.
Diseño de las sondas de captura y proceso de secuenciación
Se diseñaron sondas de captura híbrida para dichas regiones genéticas con software SureSelect® Design Tool (Agilent). Las muestras se han sometido a un proceso de extracción de ADN genómico automatizada (Qiasymphony SP®, Qiagen®). La preparación del ADN genómico se ha realizado mediante el protocolo SureSelect XT® (Agilent Technologies®), en combinación con un panel de sondas SureSelect Custom Target Enrichment®, que captura selectivamente las regiones genómicas de interés. La secuencia se realizó con equipos Illumina HiSeq 1500®. Todos los procedimientos se han llevado a cabo siguiendo las especificaciones de los fabricantes9-11.
Análisis e interpretación de datos
Se empleó un algoritmo de análisis bioinformático de desarrollo propio que permite la anotación de variantes de tipo SNP (del inglés: single nucleotide polymorphism), INDEL (inserciones-deleciones) y variantes estructurales o CNV, estas últimas mediante el análisis comparativo de la profundidad de lecturas12-14. Las variantes identificadas se combinan con el sistema de gestión del conocimiento de desarrollo propio que asocia a cada variante valores específicos de calidad e información, datos estructurales y de SNP como dbSNP, 1000 genomas15 EVS, así como datos de frecuencia de los alelos en distintas poblaciones, o valores de predictores bioinformáticos como SIFT, PMut, CADD (Tabla 1)12-14. A partir de las fuentes de información (Tabla 1), especialmente PharmVar y PharmGKB, se desarrollaron tablas en el sistema de gestión del conocimiento para realizar una inferencia automatizada de haplotipos basadas en datos de variantes tipo SNP y pequeñas INDEL. La inferencia de haplotipos de CYP2D6 se realiza con un módulo que filtra los alineamientos procedentes de CYP2D7 y CYP2D8; este filtrado tiene el objeto de evitar artefactos en las gráficas de cobertura procedentes de las lecturas en regiones homólogas16. Además, este módulo normaliza los datos de profundidad de lecturas con una muestra control con un número de copias CYP2D6 conocido y secuenciada en la misma placa de análisis. Esta normalización tiene el objeto de evitar artefactos debido a la presencia de CNV en otras muestras secuenciadas en la misma placa17. La inferencia de alelos HLA-B se realiza con un módulo específico basado en el desarrollo de Wittig et al.8.
Validación analítica de los resultados con materiales de referencia
La determinación de SNP y pequeñas INDEL (< 20 pb) se validó con la muestra Coriell® NA12878. La muestra se procesó por triplicado y los resultados obtenidos se compararon con los datos de referencia disponibles para este material resultantes de la integración de varios conjuntos de datos procedentes de la secuenciación del genoma completo en diferentes plataformas de secuenciación masiva18. Mediante este análisis se obtuvieron los valores medianos de sensibilidad (Se) y especificidad (Sp) analíticas, así como los valores predictivos positivo (VPP) y negativo (VPN). Se realizó este análisis con tres filtros diferentes de calidad usando dos parámetros de calidad NGS “Qual” (del inglés: quality score) y “DP” (del inglés: sequencing depth o profundidad de lecturas): alta (Qual > 49 & DP > 29), intermedia (Qual > 49 & DP > 14), baja (Qual > 0 & DP > 0)19.
La determinación de haplotipos farmacogenéticos basados en SNP y pequeñas INDEL se validó con la muestra Coriell® NA12878. Los resultados obtenidos se compararon con los datos de referencia para este material resultantes de la integración de varios conjuntos de datos generados con distintas plataformas de farmacogenética (proyecto GeT-RM)20,21. Estos datos incluyen los siguientes genes: CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2E1, CYP3A4, CYP3A5, CYP4F2, DPYD, GSTM1, GSTP1, GSTT1, NAT1, NAT2, SLC15A2, SLC22A2, SLCO1B1, SLCO2B1, TPMT, UGT1A1, UGT2B7, UGT2B15, UGT2B17, VKORC1. HLA-B
y CYP2D6, por su complejidad, se incluyeron un mayor número de probandos, las muestras Coriell® NA12878, NA02016, NA17254 y NA17281, que incluyen distintos tipos de variantes estructurales de CYP2D6 y alelos HLA-B de interés clínico (HLA-B*58:01)8,20,21. Mediante este análisis se obtuvo la concordancia entre los haplotipos determinados y los haplotipos de referencia.
Resultados
Regiones genéticas de interés incluidas en el panel
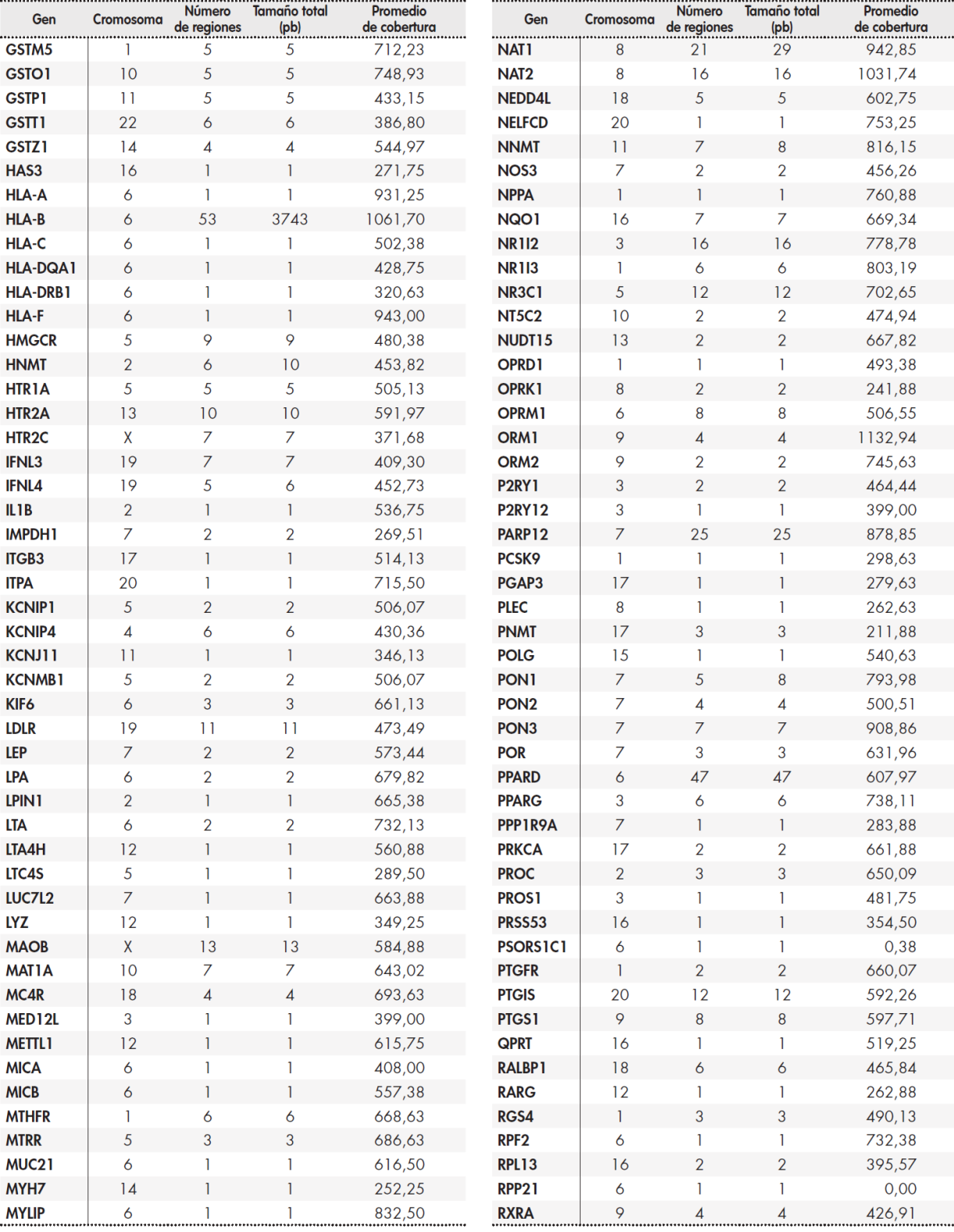
El panel desarrollado incluye un total de 12.794 pares de bases comprendidas en 389 genes. En la Tabla Suplementaria Tabla 13, Tabla 14, Tabla 15, Tabla 16 se incluyen el tamaño de la región en pares de bases que se estudia para cada gen y la cobertura promedio de la región. Estas regiones se clasificaron en tres grupos: genes principales, secundarios y candidatos, siguiendo los criterios internos que se detallan a continuación:
Genes principales (17): CYP2C19, CYP2C9, CYP2D6, CYP3A5, CYP4F2, DPYD, F5, G6PD, HLA-B, IFNL3, RARG, SLC28A3, SLCO1B1, TPMT, UGT1A1, UGT1A6, VKORC1. Son genes descritos en las guías de práctica clínica elaboradas por los principales consorcios clínicos de farmacogenética (Tabla 1). Por lo tanto, cuentan con los niveles de evidencia más altos y para todos ellos se han publicado estrategias de dosificación personalizada en base al genotipo.
Genes secundarios (50): ABCB1, ABCC2, ABCG2, ACE, ADH1A, ADH1B, ADH1C, ADRB1, ADRB2, AHR, ALDH1A1, ALOX5, ASS1, COMT, CPS1, CYP1A1, CYP1A2, CYP2A6, CYP2C8, CYP2E1, CYP2J2, CYP3A4, DRD2, GSTM1, GSTP1, GSTT1, HMGCR, KCNJ11, MTHFR, NAT1, NAT2, NQO1, NR1I2, P2RY1, P2RY12, POLG, PTGIS, SCN1A, SLC15A2, SLC19A1, SLC22A1, SLC22A6, SLC6A4, SLCO1B3, SULT1A1, TYMS, UGT2B15,
UGT2B17, UGT2B7, VDR. Los genes secundarios, a pesar de no estar incluidos en guías de práctica clínica, consideramos que pueden tener interés clínico según las fuentes de datos consultadas (Tabla 1). En este grupo destacan los listados de biomarcadores farmacogenéticos de la Food and Drug Administration, genes PharmGKB con niveles de evidencia de 2 o superior, categorías A y B de prioridad CPIC o genes core del listado pharmaADME. Aunque no se han publicado estrategias para la individualización del tratamiento en base el genotipo, consideramos que la presencia de variantes en estos genes puede ser de utilidad para la evaluación de la respuesta al tratamiento en pacientes individuales tras un hallazgo clínico.
Genes candidatos: corresponde a regiones genómicas incluidas en alguna de las principales plataformas farmacogenómicas empleadas habitualmente en la práctica clínica22,23, o regiones descritas clasificadas en las fuentes de información empleadas (Tabla 1) con niveles de evidencia inferiores a los otros grupos, por ejemplo, PharmGKB nivel 3 o 4, CPIC C/D. Su estudio se contempla en un contexto de investigación. El listado completo de genes y regiones se encuentra en el suplemento del artículo.
Validación de la determinación de SNP y pequeñas INDEL
Los resultados de la determinación de variantes genéticas de tipo sustituciones puntuales (SNP) y pequeñas inserciones/deleciones (INDEL) ≤ 20 pb se muestran en la Tabla 2. La sensibilidad y especificidad analíticas del ensayo fueron superiores al 99%. Por otro lado, los VPP y VPN fueron superiores también al 99% para todos los valores de calidad analizados.
Tabla 2. Validación de la determinación de variantes genéticas de tipo sustituciones puntuales (SNP) y pequeñas inserciones/deleciones (INDEL) < 20 pb
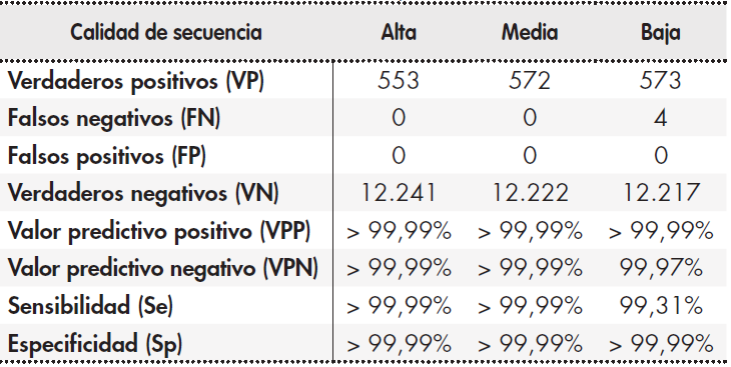
La muestra NA12878 se proceso por triplicado y los resultados se compararon con los datos de referencia generados en diferentes plataformas de secuenciacion masiva18; se muestra la mediana de los valores para las tres ejecuciones. Los filtros de calidad de secuencia se definieron como alta (Qual > 49 and DP > 29), intermedia (Qual > 49 and DP > 14) o baja (Qual > 0 amd DP > 0).
Validación de la determinación de alelos farmacogenéticos (haplotipos)
Los resultados de la determinación de haplotipos farmacogenéticos con la muestra Coriell® NA12878 se muestra en la Tabla 3. Se dispone de datos de referencia para 28 genes (cada uno con 2 haplotipos, 56 haplotipos)(20.21); los resultados fueron diferentes en cuatro genes: CYP2D6, NAT2, SLCO1B1 y UGT2B17. Para los genes CYP2D6, NAT2 y SLCO1B1 las diferencias fueron debidas a que la plataforma NGS desarrollada posee un número mayor de regiones genéticas y por lo tanto supera las capacidades de detección de las plataformas empleadas en el proyecto GeT-RM(20.21). Para CYP2D6, la plataforma NGS desarrollada detectó la presencia de un reordenamiento de tipo híbrido D6/D7 en tándem, descrito en publicaciones anteriores24,25. Para NAT2 la técnica fue capaz de detectar el alelo *12, este alelo sólo está incluido en la plataforma Agena Bioscience iPLEX ADME PGx Pro®. Para SLCO1B1 nuestra técnica identificó dos combinaciones posibles de haplotipos *1A/*15 y *1B/*5; este resultado es similar al que determina la plataforma Affimetrix DMET® y se debe a la inclusión de los subalelos *1A y *1B. Para el caso de UGT2B17 nuestra técnica no detectó la presencia del haplotipo *2, que corresponde con una deleción del gen. Este requisito no se incluyó en las especificaciones del diseño. Teniendo esto en cuenta, la concordancia general en la determinación de alelos es superior al 98%. Si el análisis se restringe a aquellos genes incluidos en las principales guías de práctica clínica (Tabla 1), la concordancia se eleva hasta el 100%.
Tabla 3. Validación de la determinación de alelos farmacogenéticos (haplotipos)
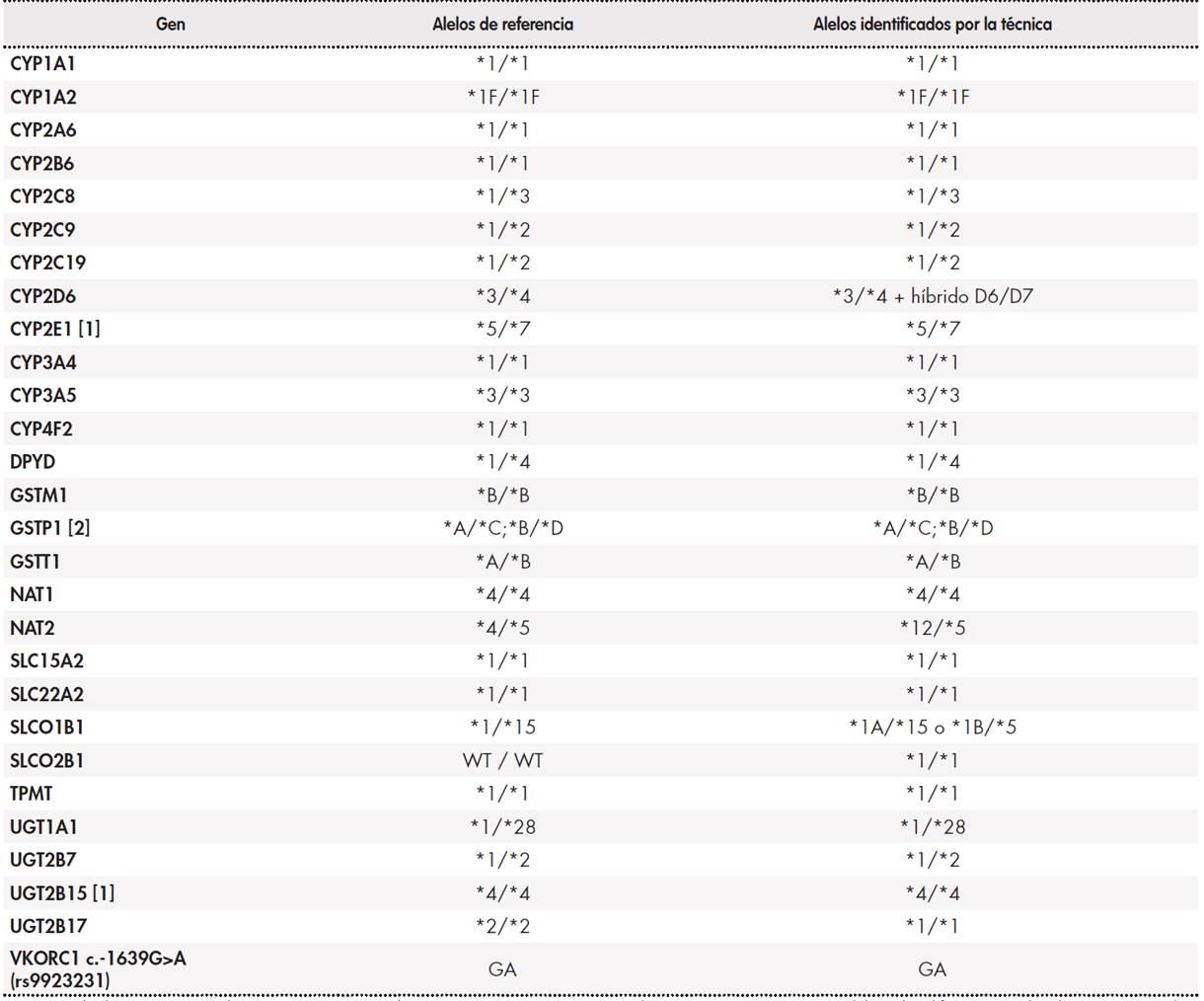
(1) Los resultados GeT-RM para la muestra NA12878 y los genes CYP2E1 y UGT2B15 indican “no consensus”. Esto se debe a las diferencias en las dos tecnicas empleadas: Affimetrix DMETR y Agena Bioscience iPLEXR. El estudio iPLEXR no es capaz de detectar el alelo CYP2E1*4 ni el UGT2B15*5. Por lo tanto, en la tabla se incluyen los resultados de Affimetrix DMETR. (2) Para GSTP1 el resultado consenso es ambiguo, esto es, se establecen dos combinaciones de alelos posibles para una combinacion de variantes determinada
Validación de los resultados de la identificación de variantes estructurales en CYP2D6
La técnica basada en el estudio comparativo de profundidad de lecturas desarrollada fue capaz de identificar adecuadamente las variantes estructurales presentes en las cuatro muestras control seleccionadas. Los resultados del análisis gráfico de coberturas para CYP2D6 se muestran en la Figura 1.

 La muestra NA17254 con el genotipo CYP2D6*4/*41 presenta dos alelos de CYP2D6, se trata de la muestra control frente a la que se normaliza la profundidad de lecturas de CYP2D6. NA17281 con el genotipo CYP2D6*5/*9 presenta una deleción de uno de los alelos CYP2D6, la profundidad de lecturas normalizada tuvo decremento de aproximadamente un 50%. NA02016 con el genotipo CYP2D6*2x2/*17 tiene una duplicación de uno de los alelos, tuvo un incremento del 50% en la profundidad de lecturas. La muestra NA12878 con el genotipo*3/*4+68 tiene un híbrido D6/D7 en tándem, tuvo un incremento de 50% en la profundidad de lecturas en una parte de la región de CYP2D6 y una parte de la región de CYP2D7.
La muestra NA17254 con el genotipo CYP2D6*4/*41 presenta dos alelos de CYP2D6, se trata de la muestra control frente a la que se normaliza la profundidad de lecturas de CYP2D6. NA17281 con el genotipo CYP2D6*5/*9 presenta una deleción de uno de los alelos CYP2D6, la profundidad de lecturas normalizada tuvo decremento de aproximadamente un 50%. NA02016 con el genotipo CYP2D6*2x2/*17 tiene una duplicación de uno de los alelos, tuvo un incremento del 50% en la profundidad de lecturas. La muestra NA12878 con el genotipo*3/*4+68 tiene un híbrido D6/D7 en tándem, tuvo un incremento de 50% en la profundidad de lecturas en una parte de la región de CYP2D6 y una parte de la región de CYP2D7.
Validación de los resultados de la identificación de variantes en HLA
La técnica desarrollada para la imputación de los alelos en la región de HLA-B fue capaz de identificar adecuadamente los alelos presentes en las tres muestras control seleccionadas. La Tabla 4 muestra los alelos consenso establecidos y los resultados obtenidos por nuestra técnica.

Discusión
En este artículo se describe el desarrollo y la validación analítica de una plataforma de secuenciación NGS para la farmacogenética clínica, así como los resultados obtenidos con las herramientas bioinformáticas empleadas en la interpretación de los datos.
El panel desarrollado cubre un total de 12.794 pares de bases comprendidas en 389 genes. Este tamaño es muy superior al de las plataformas convencionales más exhaustivas que se emplean habitualmente: Affymetrix DMET® (1.936 marcadores), Agena Bioscience® (Sequenom®), iPLEX ADME PGx Pro® (270 marcadores), Illumina VeraCode ADME® (184 marcadores) y Roche Amplichip® (36 alelos CYP2D6 y CYP2C19)22,23.
Los resultados obtenidos en las muestras control fueron coherentes con los resultados de referencia del proyecto GeT-RM. Las diferencias obtenidas se deben a una capacidad superior de nuestra plataforma, salvo en el caso de UGT2B7. Para este gen se han descrito variantes estructurales de tipo deleción que requería un diseño específico en nuestra plataforma. Dicho diseño se descartó en las especificaciones, ya que no ha sido incluido en ninguna de las principales guías de práctica clínica farmacogenéticas. Ciertas regiones genómicas de gran interés farmacogenético como CYP2D6 y HLA-B son complejas de estudiar. CYP2D6 es responsable del metabolismo de aproximadamente un 25% de los fármacos comercializados26 y su genotipo es esencial para individualizar tratamientos antidepresivos como paroxetine27, antipsicóticos como aripiprazole28, tamoxifeno29, antieméticos como ondansetron30 o analgésico como codeína31, entre otros. La dificultad para su análisis genético radica en la existencia de dos regiones con muy alta homología, CYP2D7 y CYP2D8, y a la existencia de variantes tipo CNV y reordenamientos con los pseudogenes (híbridos de tipo conversión y en tándem con alelos funcionales). Se han publicado otras herramientas para el análisis bioinformático de CYP2D6 que obtienen buenos resultados en el análisis de variantes tipo SNP o pequeñas INDEL, pero con limitaciones en la determinación de variantes estructurales, especialmente híbridos16,17,24,32,34. Para este fin, tal y como se expone en la sección de materiales y métodos, la técnica NGS desarrollada realiza un filtrado de lecturas procedentes de las regiones homólogas y normaliza la profundidad de lecturas frente a una muestra control conocida. La plataforma NGS desarrollada ha identificado de manera adecuada diferentes tipos de variantes estructurales y CNV analizados en materiales de referencia y proporciona unas gráficas con la profundidad de lecturas que muestran el comportamiento de la cobertura a lo largo de toda la región genómica (Figura 1).
La región HLA (antígeno leucocitario humano) es la región más densamente polimórfica del genoma. Su importancia farmacogenética radica en su asociación con las reacciones de hipersensibilidad a fármacos inmuno-mediadas34. Las técnicas de tipado convencionales permiten solamente el análisis de unos pocos haplotipos, mientras que los procedimientos generales para el estudio de dicha región mediante tecnologías NGS no obtienen buenos resultados debido a la variabilidad y la homología de secuencia entre los genes y los pseudogenes35. El presente desarrollo NGS incorpora un diseño de sondas específico y un software de análisis previamente validado que permite un estudio global de HLA-B.
Las principales limitaciones de la plataforma NGS en comparación con las plataformas convencionales incluyen el tiempo de estudio, la cualificación del personal técnico necesario, los costes de desarrollo y puesta a punto, así como los costes de procesamiento informático y almacenamiento de la información en servidores36. El tiempo necesario para obtener el resultado desde que se recibe una muestra es de aproximadamente 10 días (incluyendo preparación de la muestra, secuenciación y análisis bioinformático). Además del equipamiento analítico y servidores para procesado de datos, estos procesos requieren de personal técnico con formación específica y personal bioinformático para la puesta a punto de la técnica y el análisis de los datos de secuenciación.
Aunque la técnica y el proceso de datos bioinformático desarrollado identifican variantes raras y otras variantes no descritas previamente, estos resultados no se trasladan a la práctica clínica asistencial, ya que no se dispone de datos en la literatura para su interpretación. Se ha descrito que más del 80% de la variación en los genes de interés en farmacogenética se debe a la presencia de estas variantes raras y las plataformas convencionales no están orientadas a su análisis37,38. Este tipo de variantes podrán incorporarse a la práctica clínica asistencial a medida que se disponga de protocolos y consensos para su interpretación, así como de publicaciones científicas con datos de correlación genotipo-fenotipo39.
Por último, para demostrar el valor clínico de esta plataforma, es conveniente su evaluación y validación en estudios clínicos prospectivos
Aportación a la literatura científica
El presente trabajo aborda el desarrollo, implementación y validación de un panel de secuenciación masiva en paralelo para la farmacogenética clínica.

Tabla suplementaria 1 (cont.). Listado de regiones genómicas, tamaño y cobertura promedio para cada gen
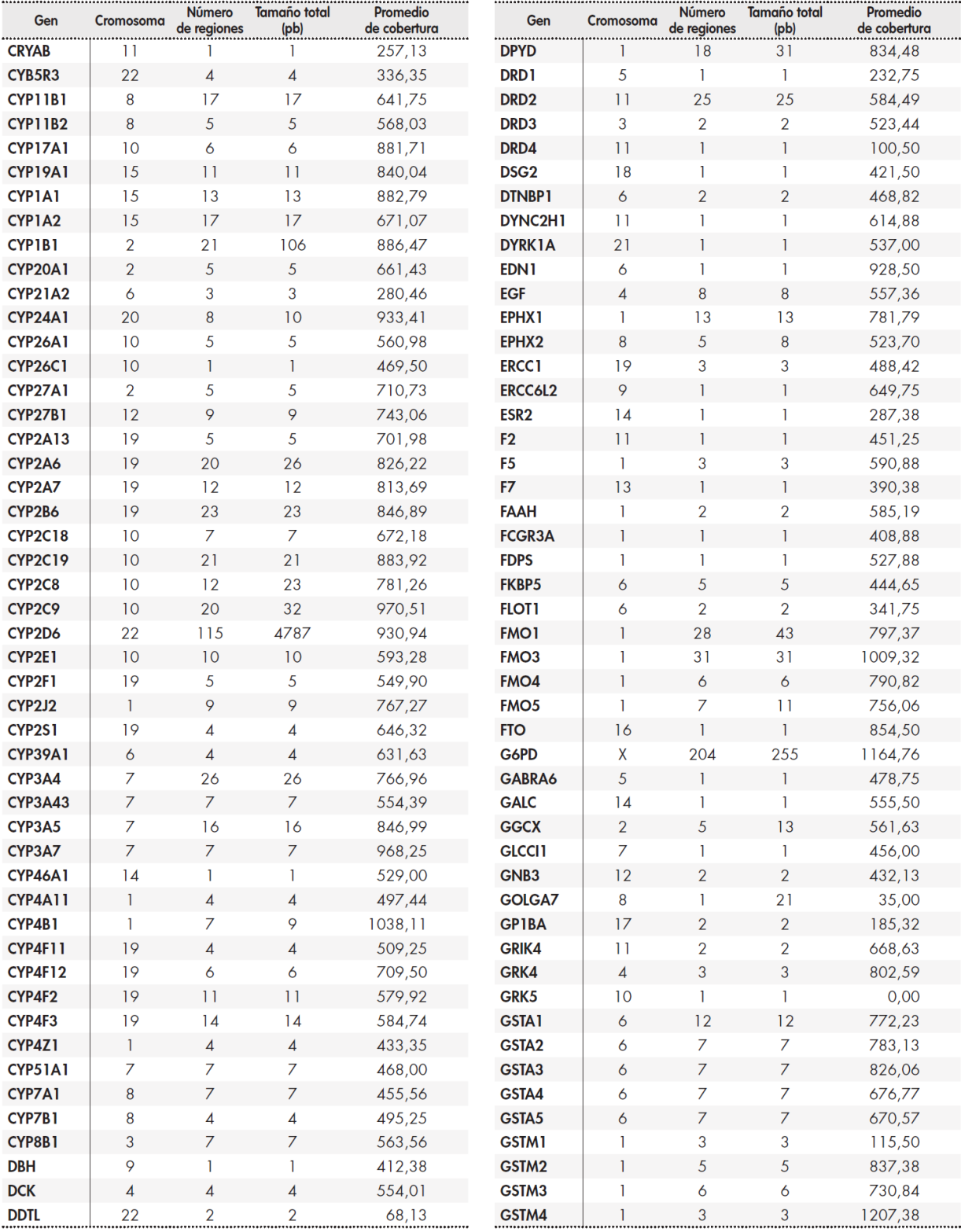
Tabla suplementaria 1 (cont.). Listado de regiones genómicas, tamaño y cobertura promedio para cada gen

Bibliography
Roden DM, McLeod HL, Relling MV, Williams MS, Mensah GA, Peterson JF, et al. Pharmacogenomics. Lancet (Internet). 2019;6736(19):1-12. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0140673619312760 [ Links ]
Han SM, Park J, Lee JH, Lee SS, Kim H, Han H, et al. Targeted Next-Generation Sequencing for Comprehensive Genetic Profiling of Pharmacogenes. Clin Pharmacol Ther. 2017;101(3):396-405. [ Links ]
Ji Y, Si Y, McMillin GA, Lyon E. Clinical pharmacogenomics testing in the era of next generation sequencing: challenges and opportunities for precision medicine. Expert Rev Mol Diagn (Internet). 2018;18(5):411-21. Available at: https://doi.org/10.1080/14737159.2018.1461561 [ Links ]
Bank P, Caudle K, Swen J, Gammal R, Whirl-Carrillo M, Klein T, et al. Comparison of the Guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin Pharmacol Ther (Internet). 2017;4457. Available at: http://onlinelibrary.wiley.com/doi/10.1002/cpt.762/full [ Links ]
Bousman CA, Jaksa P, Pantelis C. Systematic evaluation of commercial pharmacogenetic testing in psychiatry. Pharmacogenet Genomics (Internet). 2017;27(11):387-93. Available at: https://www.ncbi.nlm.nih.gov/pubmed/28777243 [ Links ]
Kalman LV, Agúndez JAG, Appell ML, Black JL, Bell GC, Boukouvala S, et al. Pharmacogenetic Allele Nomenclature: International Workgroup Recommendations for Test Result Reporting. Clin Pharmacol Ther (Internet). 2016;99(2):172-85. Available at: https://www.ncbi.nlm.nih.gov/pubmed/26479518 [ Links ]
Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE. IPD-IMGT/ HLA Database. Nucleic Acids Res (Internet). 2019 Oct 31. Available at: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkz950/5610347 [ Links ]
Wittig M, Anmarkrud JA, Kassens JC, Koch S, Forster M, Ellinghaus E, et al. Development of a high-resolution NGS-based HLA-typing and analysis pipeline. Nucleic Acids Res (Internet). 2015;43(11):e70-e70. Available at: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkv184 [ Links ]
Agilent Technologies. SureSelectXT Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library (Internet). 2019. Available at: https://www.agilent.com/cs/library/usermanuals/Public/G7530-90000.pdf [ Links ]
Qiagen. Manual de instrucciones de uso de QIAsymphony® DSP DNA (Internet). 2015. Available at: https://www.qiagen.com/fr/resources/download.aspx?id=8bc88dad-4140-467e-a1f0-e390fd193865&lang=es-ES [ Links ]
Illumina. HiSeq 1500 System Guide (15035788 v03) (Internet). 2019. Available at: https://emea.support.illumina.com/downloads/hiseq_1500_user_guide_15035788.html [ Links ]
Ochoa JP, Sabater-Molina M, García-Pinilla JM, Mogensen J, Restrepo-Córdoba A ,Palomino-Doza J, et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a Genetic Basis for Hypertrophic Cardiomyopathy. J Am Coll Cardiol (Internet). 2018;72(20): 2457-67. Available at: http://www.ncbi.nlm.nih.gov/pubmed/30442288 [ Links ]
Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol (Internet). 2016;68(22):2440-51. Available at: http://www.ncbi.nlm.nih.gov/pubmed/27908349 [ Links ]
Trujillo-Quintero JP, Gutiérrez-Agulló M, Ochoa JP, Martínez-Martínez JG, de Uña D, García-Fernández A. Familial Brugada Syndrome Associated With a Complete Deletion of the SCN5A and SCN10A Genes. Rev Esp Cardiol (Engl Ed) (Internet). 2019;72(2):176-8. Available at: http://www.ncbi.nlm.nih.gov/pubmed/29650450 [ Links ]
The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature (Internet). 2012;491(7422):56-65. Available at: http://www.nature.com/articles/nature11632 [ Links ]
Numanagić I, Malikić S, Pratt VM, Skaar TC, Flockhart DA, Sahinalp SC. Cypiripi: exact genotyping of CYP2D6 using high-throughput sequencing data. Bioinformatics (Internet). 2015;31(12):i27-34. Available at: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btv232 [ Links ]
Lee S, Wheeler MM, Patterson K, McGee S, Dalton R, Woodahl EL, et al. Stargazer: a software tool for calling star alleles from next-generation sequencing data using CYP2D6 as a model. Genet Med (Internet). 2019;21(2):361-72. Available at: http://dx.doi.org/10.1038/s41436-018-0054-0 [ Links ]
Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol (Internet). 2014;32(3):246-51. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24531798 [ Links ]
BCFtools - Variant Calling (Internet) (accessed 06/08/2020). Available at: https://samtools.github.io/bcftools/howtos/variant-calling.html [ Links ]
Pratt VM, Everts RE, Aggarwal P, Beyer BN, Broeckel U, Epstein-Baak R, et al. Characterization of 137 Genomic DNA Reference Materials for 28 Pharmacogenetic Genes: A GeT-RM Collaborative Project. J Mol Diagnostics (Internet). 2016;18(1):109-23. Available at: http://dx.doi.org/10.1016/j.jmoldx.2015.08.005 [ Links ]
Pratt VM, Zehnbauer B, Wilson JA, Baak R, Babic N, Bettinotti M, et al. Characterization of 107 Genomic DNA Reference Materials for CYP2D6, CYP2C19, CYP2C9, VKORC1, and UGT1A1. J Mol Diagnostics (Internet). 2010;12(6):835-46. Available at: https://linkinghub.elsevier.com/retrieve/pii/S1525157810601341 [ Links ]
Arbitrio M, Di Martino MT, Scionti F, Agapito G, Hiram Guzzi P, Cannataro M, et al. DMETTM (Drug Metabolism Enzymes and Transporters): a Pharmacogenomic platform for precision medicine. Oncotarget (Internet). 2016;5(0). Available at: http://www.oncotarget.com/abstract/9927 [ Links ]
Agena Bioscience: iPLEX® PGx Pro Panel Flyer (Internet) (accessed 06/04/2020). Available at: https://agenabio.com/resources/product-literature/ [ Links ]
Twist GP, Gaedigk A, Miller NA, Farrow EG, Willig LK, Dinwiddie DL, et al. Constellation: a tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole-genome sequences. npj Genomic Med (Internet). 2016;1(August 2015):15007. Available at: http://www.nature.com/articles/npjgenmed20157 [ Links ]
Gaedigk A, Turner A, Everts RE, Scott SA, Aggarwal P, Broeckel U, et al. Characterization of Reference Materials for Genetic Testing of CYP2D6 Alleles: A GeT-RM Collaborative Project. J Mol Diagnostics (Internet). 2019;21(6):1034-52. Available at: https://doi.org/10.1016/j.jmoldx.2019.06.007 [ Links ]
Owen RP, Sangkuhl K, Klein TE, Altman RB. Cytochrome P450 2D6. Pharmacogenet Genomics (Internet). 2009;19(7):559-62. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19512959 [ Links ]
Hicks JK, Bishop JR, Sangkuhl K, Muller DJ, Ji Y, Leckband SG, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-34. [ Links ]
Swen JJ, Nijenhuis M, de Boer A, Grandia L, Maitland-van der Zee AH, Mulder H, et al. Pharmacogenetics: from bench to byte--an update of guidelines. Clin Pharmacol Ther (Internet). 2011;89(5):662-73. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21412232 [ Links ]
Drögemöller BI, Wright GEB, Shih J, Monzon JG, Gelmon KA, Ross CJD, et al. CYP2D6 as a treatment decision aid for ER-positive non-metastatic breast cancer patients: a systematic review with accompanying clinical practice guidelines. Breast Cancer Res Treat (Internet). 2019;173(3):521-32. Available at: http://dx.doi.org/10.1007/s10549-018-5027-0 [ Links ]
Bell GC, Caudle KE, Whirl-Carrillo M, Gordon RJ, Hikino K, Prows CA, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 genotype and use of ondansetron and tropisetron. Clin Pharmacol Ther (Internet). 2017;102(2):213-8. Available at: http://www.pharmgkb.org/page/cpic/ [ Links ]
Madadi P, Amstutz U, Rieder M, Ito S, Fung V, Hwang S, et al. Clinical practice guideline: CYP2D6 genotyping for safe and efficacious codeine therapy. J Popul Ther Clin Pharmacol. 2013;20(3):369-96. [ Links ]
Klein TE, Ritchie MD. PharmCAT: A Pharmacogenomics Clinical Annotation Tool. Clin Pharmacol Ther (Internet). 2018;104(1):19-22. Available at: http://doi.wiley.com/10.1002/cpt.928 [ Links ]
Numanagić I, Malikić S, Ford M, Qin X, Toji L, Radovich M, et al. Allelic decomposition and exact genotyping of highly polymorphic and structurally variant genes. Nat Commun (Internet). 2018;9(1):828. Available at: http://www.nature.com/articles/s41467-018-03273-1 [ Links ]
Barbarino JM, Kroetz DL, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for human leukocyte antigen B. Pharmacogenet Genomics (Internet). 2015;25(4):205-21. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25647431 [ Links ]
Hosomichi K, Shiina T, Tajima A, Inoue I. The impact of next-generation sequencing technologies on HLA research. J Hum Genet (Internet). 2015;60(11):665-73. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26311539 http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4660052 [ Links ]
Roy S, Coldren C, Karunamurthy A, Kip NS, Klee EW, Lincoln SE, et al. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines. J Mol Diagnostics (Internet). 2018;20(1):4-27. Available at: https://linkinghub.elsevier.com/retrieve/pii/S1525157817303732 [ Links ]
Hovelson DH, Xue Z, Zawistowski M, Ehm MG, Harris EC, Stocker SL, et al. Characterization of ADME gene variation in 21 populations by exome sequencing. Pharmacogenet Genomics (Internet). 2017;27(3):89-100. Available at: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=01213011-900000000-99273 [ Links ]
Kozyra M, Ingelman-Sundberg M, Lauschke VM. Rare genetic variants in celular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet Med. 2017;19(1):20-9. [ Links ]
Lauschke VM, Ingelman-Sundberg M. How to Consider Rare Genetic Variants in Personalized Drug Therapy. Clin Pharmacol Ther (Internet). 2018;103(5):745-8. Available at: http://dx.doi.org/10.1002/cpt.976 [ Links ]
Recibido: 12 de Abril de 2020; Aprobado: 13 de Junio de 2020
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
