










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCTION
Breakthrough pain is a transitory flare of pain that occurs on a background of relatively well controlled baseline pain and is highly prevalent among patients with cancer. Breakthrough cancer pain (BTCP) shows high inter- and intraindividual variability in rate of onset, maximum intensity, time to maximum intensity and duration 1,2,3.
Opioids are the mainstay of cancer pain pharmacological treatment, but oral opioids can poorly adapt to the rapid onset and short duration of BTCP, which prompted the development of new products that could fit better with this specific time-course of effects, optimizing the balance between pain relief and side-effects (3,4,5).
Oral transmucosal fentanyl citrate (OTFC), a formulation of fentanyl citrate embedded in a sweetened lozenge on a plastic handle (stick), was the first product specifically designed for the treatment of BTCP. Oral transmucosal absorption of fentanyl provides with greater and faster bioavailability than enteral formulations, thus allowing a faster pain relief 4,5,6.
Since its approval, several other oral or nasal transmucosal absorption formulations have been approved. They show some differences in their pharmacokinetic profile, and some recommendations point to selecting the product that best matches with the individual characteristics of each patient and pain episode. OTFC owes a characteristic unique among all fentanyl transmucosal products: it is self-administered through a dynamic process that the patient can control to achieve the desired effects, interrupting the administration if pain relief or side-effects occur. This unique feature is termed modulability, flexibility or self-control 2,4,7.
The present study aimed at proving bioequivalence of a test OTFC product compared to the reference formulation.
METHODS
A phase 1, open-label, crossover, single-dose bioequivalence study with two study periods and two sequences comparing the bioavailability of 2 OTFC formulations was conducted at the Clínica Universidad de Navarra Clinical Research Unit in accordance with the "Note for Guidance on Good Clinical Practice" (CPMP/ICH/135/95) 8, the "Guidelines of the Investigation of Bioequivalence" (CPMP/EWP/QWP/1401/98 Rev. 1) 9, and the Declaration of Helsinki (revision, Seoul, 2008). The protocol was approved by the Independent Ethics Committee of Navarre and the volunteers signed their written consent before participating.
Subjects
Participants have to be healthy volunteers of both sexes, aged between 18 and 45 years old, non-smokers, with a body mass index between 19 and 29 kg/m2, and with an oxygen saturation equal to or greater than 95 %. Each volunteer underwent an anamnesis, a physical examination, an ECG and analysis before being included in the study to rule out any type of disease.
Design
The duration of the study was 59 days, divided into three phases. During the first phase (screening phase, 21 days), the suitability of the volunteers was evaluated according to the inclusion and exclusion criteria and they underwent a medicinal product administration training process in order to achieve a technique the most accurate and homogeneous possible in all participants. The second phase (intervention phase) involved the two treatment periods, separated by a washout period of at least 10 days, during which the volunteer received, while fasting and at random, one of two formulations: 400 μg of fentanyl Geiser Pharma (test) or Actiq 400 μg (reference, Cephalon UK Ltd.). Holding the product by its handle, volunteers had to place the fentanyl tablet in their mouth onto the interior side of the cheek and rub it gently against the buccal mucosa, moving it around and rotating the tablet, in order to maximize the mucosal exposure of fentanyl. They were also reminded that they should neither suck on nor chew the tablet. The investigators controlled each administration to homogenize it in all subjects, ensuring that it was consumed during 15 minutes as indicated in the Summary of Product Characteristics for the product 10. Beforehand, and to prevent adverse reactions of fentanyl (especially respiratory depression), 50 mg of naltrexone antidote was administered 12 hours prior, immediately before taking fentanyl, and 12 hours afterwards. After the administration of the drug, blood draws were taken at the following times: 0, 5, 10, 15, 20, 30, 40, 50 minutes and at 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 24, 48 and 72 hours, to determine the pharmacokinetic parameters studied. The safety evaluation was carried out with the measurement of blood pressure (BP), heart rate (HR), respiratory rate (RR), temperature (T) and O2 saturation prior to administration of the drug and at 15, 20, 30, 40, 50 minutes and at 1, 1.5, 2, 2.5, 3, 4 and 8 hours. They were also asked about the onset of adverse events (AEs) after each blood draw. The volunteers remained in hospital from 12 hours before until 12 hours after the administration of the drug. The final phase (follow-up) was conducted during the week posterior to the administration of the second drug dose. It consisted of a physical examination (weight, RR, T, BP, HR and pulse oximetry), a 12-lead ECG and a complete blood test.
Given the variability of the pharmacokinetic parameters described in the literature with this formulation 5, a two-stage design was established allowing a sample size reestimation for a second stage based on the variance estimated from the first stage, if necessary 9. In the first one, 36 subjects were initially included and a first analysis was performed with the data obtained, in such a way that if it was concluded that both formulations were BE, the study would be stopped; otherwise, the intraindividual variability observed would be used for the definitive calculation of volunteers for the second stage, which would be at least 12 more volunteers.
Pharmacokinetic analysis
The determination of fentanyl in plasma was performed by high-performance liquid chromatography with mass spectrometry/(HPLC/MS/MS) using a validated method. The linear relationship between the detector response and the plasma fentanyl concentrations was checked throughout the range of concentrations between 20-5,000 pg/ml. Sample handling included blood draw with a tube with a EDTA K2 anticoagulant, centrifugation at 3,000 rpm at 4 °C for 10 minutes and subsequent freezing at -35 °C for the first 24 hours and at -80 °C the following hours, until transferred to the analytical laboratory.
The following pharmacokinetic parameters of fentanyl were calculated in each subject after the administration of each formulation: Cmax, AUC0-t, AUC0-∞, tmax and t1/2. The AUC was calculated using the linear trapezoidal method. For the bioequivalence study between the two formulations, the Cmax and AUC0-t parameters were compared after their logarithmic transformation and the parametric symmetric confidence intervals (CI) of 94.12 % were defined for each value, according to the rules stablished for two step designs in the Guidelines of the Investigation of Bioequivalence stated by the European Medicines Agency 9. To calculate the limits of this interval, a 3-way repeated measure ANOVA was applied: formulation (2 categories), sequence (2 categories) and administration period (2 categories). The two treatments were considered bioequivalent if the CI limits calculated fell within the acceptance range of 0.8-1.25 9.
The safety variables were analysed using Student's t-test for paired data or ANOVA according to each case. If the conditions for carrying out these tests were not met, the corresponding parametric tests were performed (Wilcoxon, Friedman).
RESULTS
A total of 37 volunteers were included (19 men and 18 women; mean age: 22.7 ± 4.5 years (range 18-43 years); weight: 68.7 ± 11.7 kg (50-93 kg); height: 1.7 ± 0.1 m (1.6-1.9 m); BMI: 23.3 ± 2.3 kg/m2 (19-29 kg/m2), of which 36 completed the study according to the protocol. The mean consumption time of the reference drug was 14 ± 3 minutes (range: 9-23 minutes), and that of the test drug was 15 ± 3 minutes (10-23 minutes).
Pharmacokinetic parameters
The plasma concentration of the fentanyl formulations after the administration of 400 μ is shown in Figure 1 and the pharmacokinetic parameters (Cmax, AUC0-t, AUC0-∞, tmax and t1/2) in Table I.

Fig. 1 Mean ± standard error of fentanyl plasma concentration versus time after single doses of 400 µg of Fentanyl Geiser Pharma (test) compared to 400 µg of Actiq (reference) in healthy adult volunteers (n = 36) for the first 48 h (A) and for the rapid absorption phase (B). At 72 h only 1 quantifiable sample was found (subject 05, test formulation, 23.7 pg/ml) and is not represented.
Table I Pharmacokinetic parameters (mean ± standard deviation) after single doses of 400 μg of Fentanyl Geiser Pharma (test) compared to 400 μg of Actiq (reference) in healthy adult volunteers (n = 36)

Cmax: maximum fentanyl concentration. Tmax: Time when Cmax occurs. AUC0-t: Area under the curve, calculated from time 0 to the last measured concentration. AUC0-∞: Area under the curve from time 0 extrapolated to infinite time. T1/2: half-life. *Median and range.
The limits of the confidence interval (94.12 %) of the transformed data from the Cmax and AUC0-t of fentanyl parameters fell within the theoretical bioequivalence acceptance interval, therefore both products can be considered bioequivalent (Table II). The analysis of this first stage showed an adequate statistical power to conclude in the acceptance of bioequivalence (p > 0.97) with the two evaluated parameters, Cmax and AUC0-t. The estimated intraindividual variability was 24.66 % and 19.01 %, respectively. This BE was also observed when the calculation was performed based on the classic CIs of 90 %. No period or sequence effects were observed in any parameter. Since this BE was observed in the first phase of the study, it was not necessary to extend the sample with a second stage.
Table II Bioequivalence analysis (Cmax, AUC0-t) after single doses of 400 μg of Fentanyl Geiser Pharma (test) compared to 400 μg of Actiq (reference) in healthy adult volunteers (n = 36)
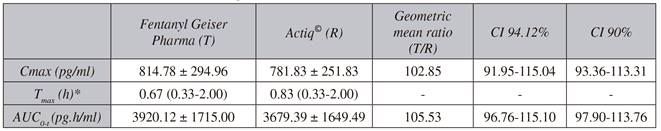
Values are mean ± standard deviation. Cmax: maximum fentanyl concentration; Tmax: Time when Cmax occurs; AUC0-t: Area under the curve, calculated from time 0 to the last measured concentration. CI: Confidence Interval. *Median and range.
Safety
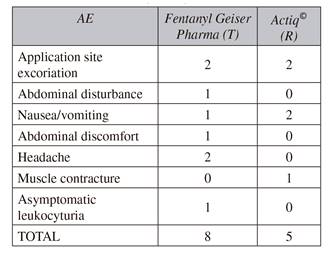
Both formulations were well tolerated. Overall, 15 adverse events were reported (eight related to the test product and 5 to the reference product), all of which were mild, 7 of these being related to the medication (Table III). All AEs were transient, although rescue medication was required in five cases. In the final check-up, no alterations were observed in any volunteer during the physical examination, ECG or hematological and biochemical test.
DISCUSSION
These results showed that both OTFC formulations are bioequivalent and therefore interchangeable when used clinically. As established in the European guidelines on bioavailability and bioequivalence (CPMP/EWP/QWP/1401/98), a similar clinical effect can be established for both formulations, without the need for the corroboration of a clinical study, as it is commonly accepted that a plasma concentration of a similar active substance is essentially achieved in the same subject in the same time 9. In our case, the geometric mean ratio (test/reference) was consistent with the parameters established in these guidelines to consider both products as BE. In addition, the two formulations presented a similar tmax, with a median of 40 minutes in the case of the test formulation and 50 minutes for the reference one, indicating a similar rate of absorption, a fundamental and differentiating aspect in this type of formulation, as BTCP requires rapid onset of pain relief 1,2,3,4,5,6.
OTFC administration yields plasma concentrations that are higher and more rapidly attained than those after oral administration: fentanyl from OTFC passes partially by mucosal transport directly into the systemic circulation without undergoing enteric absorption and first pass metabolism. In this way, a bioavailability of 50% is achieved, divided equally between fast transmucosal absorption and slower gastrointestinal absorption 5. However, these fractions could variate. Stanley et al. (1989) 11 and Streisand et al. (1991) 12, in the first studies evaluating the absorption and bioavailability of OTFC in adult volunteers, remarked the profound influence oral mucosa absorption plays on the movement of fentanyl into the bloodstream. Indeed, absorption of OTFC trough oral mucosal membranes is complex and involves numerous factors. Thus, the rate of sucking and saliva production (affected by the taste and pH of the lozenge) influences the dissolution process. Moreover, it seems that the amount of saliva immediately swallowed without adequate exposure to mucosal surfaces is a critical factor in overall absorption and probably accounts for much of the inter- and intra-patient variability associated with OTFC delivery. As mentioned in the literature, the coefficient of variation of AUC0-t and Cmax has been established within a range as wide as 7-52 % after the administration of doses from 400-800 μg of OTFC 12,13,14,15,16,17. Although inter-individual variability can be reduced by the crossover design of most BE studies, the risk of intra-individual variability may persist, especially with this type of formulation 18. According to this high variability, a two-stage crossover bioequivalence (BE) study was chosen as it allows the reestimation of the second-stage sample size based on the variance estimated from the first-stage results. However, in our case, no extension was required since BE was demonstrated after the analysis of the first 36 subjects. The thorough training during the screening phase and the active supervision of the investigator during the administration of the drug were aimed to reduce the variability in transmucosal absorption, and may have been determinant in decreasing the variance estimated and, therefore, avoiding the second stage of the study. This premise is supported by the fact that the consumption time values for both formulations (mean, maximum and minimum values) were almost identical and in accordance with the approved product label (15 minutes).
As the rate of absorption of fentanyl is highly dependent on the administration technique, OTFC allows the patient to modulate or self-control it to achieve the desired effects, interrupting the administration if pain relief or side-effects occur, providing a flexibility unique among all fentanyl transmucosal products 2,4,5,8.
In the present study, median time to maximum concentrations (tmax) was 40-50 minutes for the test and the reference formulations, respectively, which is in accordance with those originally reported for the reference product: 20-62 min 13,14,15,16,17.
Conversely, later studies reported higher figures (90-120 minutes), that indicate a slower rate of absorption 5. These differences put in evidence the influence of the technique of administration on the rate of bioavailability of fentanyl, which is determinant for its pharmacokinetic profile. In fact, the Cmax/AUC ratios of the reference formulation original studies (weighted mean 0.136; range: 0.10-0.21) are 16 % higher compared to the later studies (weighted mean 0.117; range: 0.09-0.13), which confirms a decrease in fentanyl Cmax in the latter. In the present study, Cmax/AUC ratio resulted in 0.179. Slowing the absorption rate delays the tmax, lowers the Cmax and causes longer-lasting plasma concentrations, providing a profile of pain relief that could fit better to BTCP episodes with slower onset and longer duration. Proper education of the patient is essential to optimize the use of OTFC 5. Well-trained patients can take advantage of OTFC flexibility or modulability, gaining the empowerment of self-controlling pain relief as Ashburn (1989) reported 7.
Finally, both formulations were well tolerated. The reported adverse events were mild-moderate in intensity and self-limited in most cases. However, the use of naltrexone, an opioid antagonist, could have avoided other serious adverse events opiate-related, as respiratory depression. In any case, it is worth noting that this self-administered formulation, allows the patient to remove the drug immediately if non-tolerated adverse effects appear, unlike other presentations, such as sublingual or intranasal routes 4. This advantage could provide a greater safety in its use. Nevertheless, this action was not required in our study.
In conclusion, our results showed that Fentanyl Geiser Pharma can be considered bioequivalent to the reference product, Actiq. Both will produce the same clinical effect at the same doses within the same safety range, being, therefore, interchangeable. All of this should improve the management of breakthrough pain in oncologic patients by providing an easier access to a medicinal product of proven efficacy and safety in such sensitive condition.

